Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs)


 , ,
, ,  ,
,  and
and 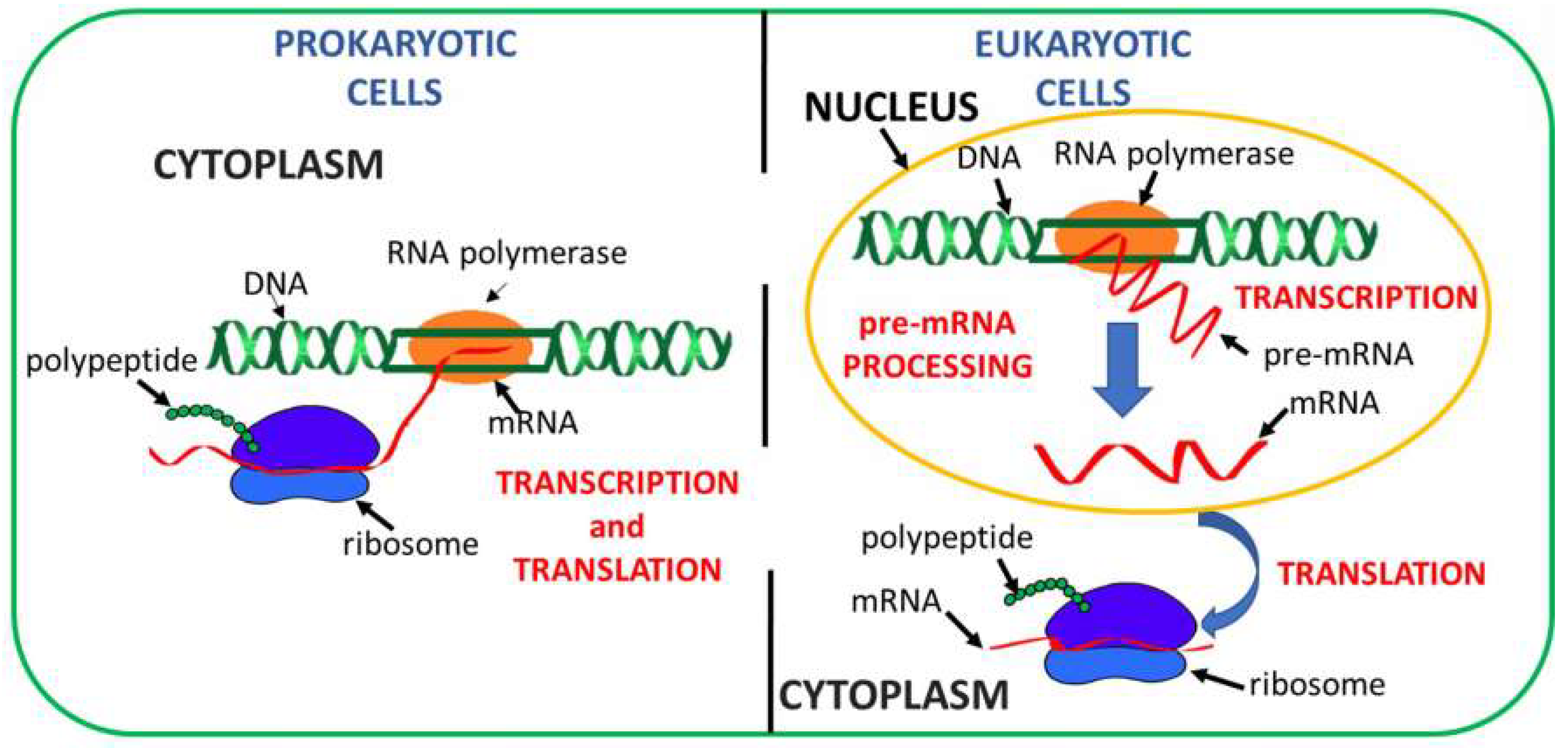{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
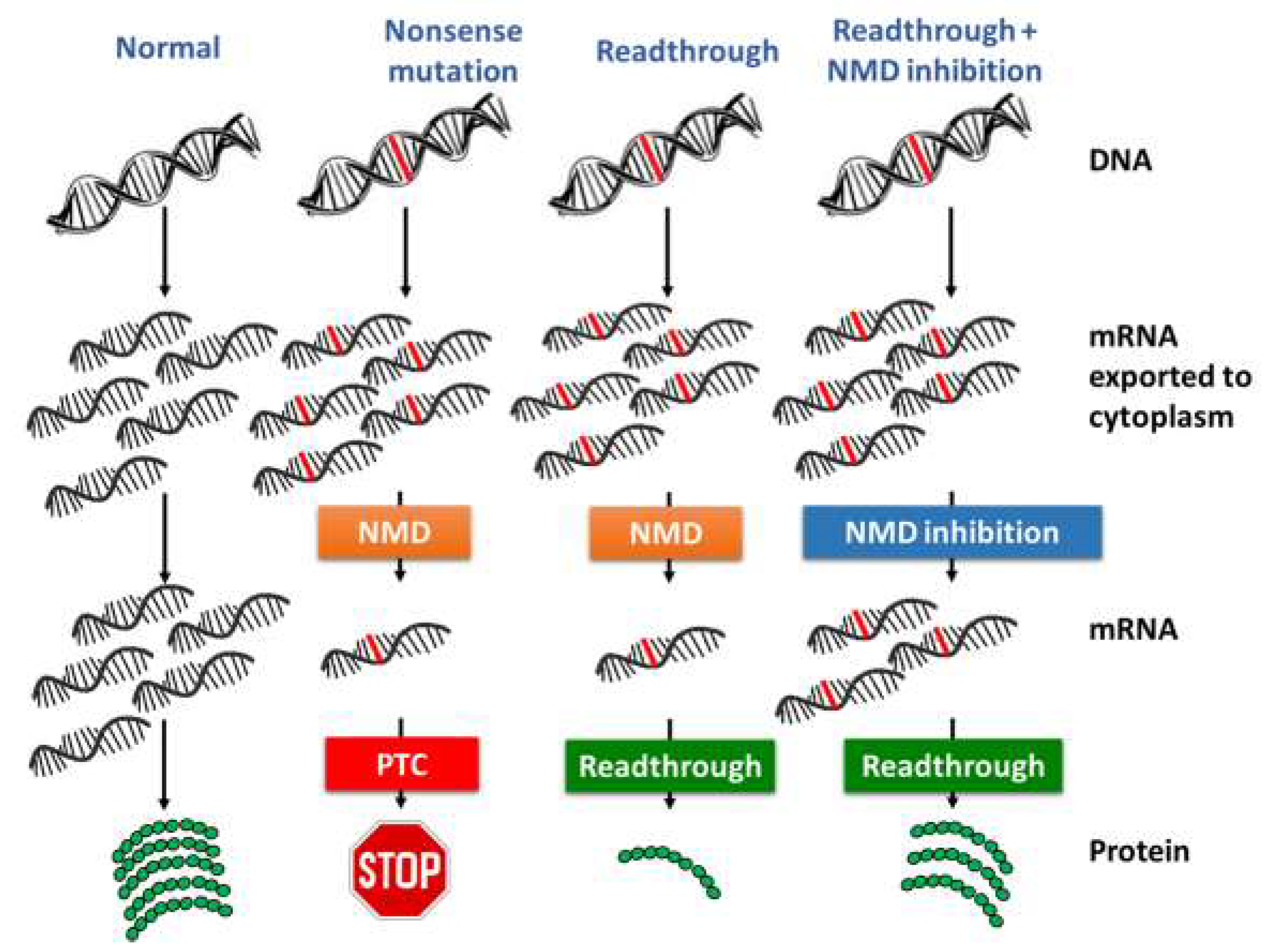
2. Concepts behind Readthrough Approaches
3. Classes of Translational Readthrough-Inducing Drugs (TRIDs)
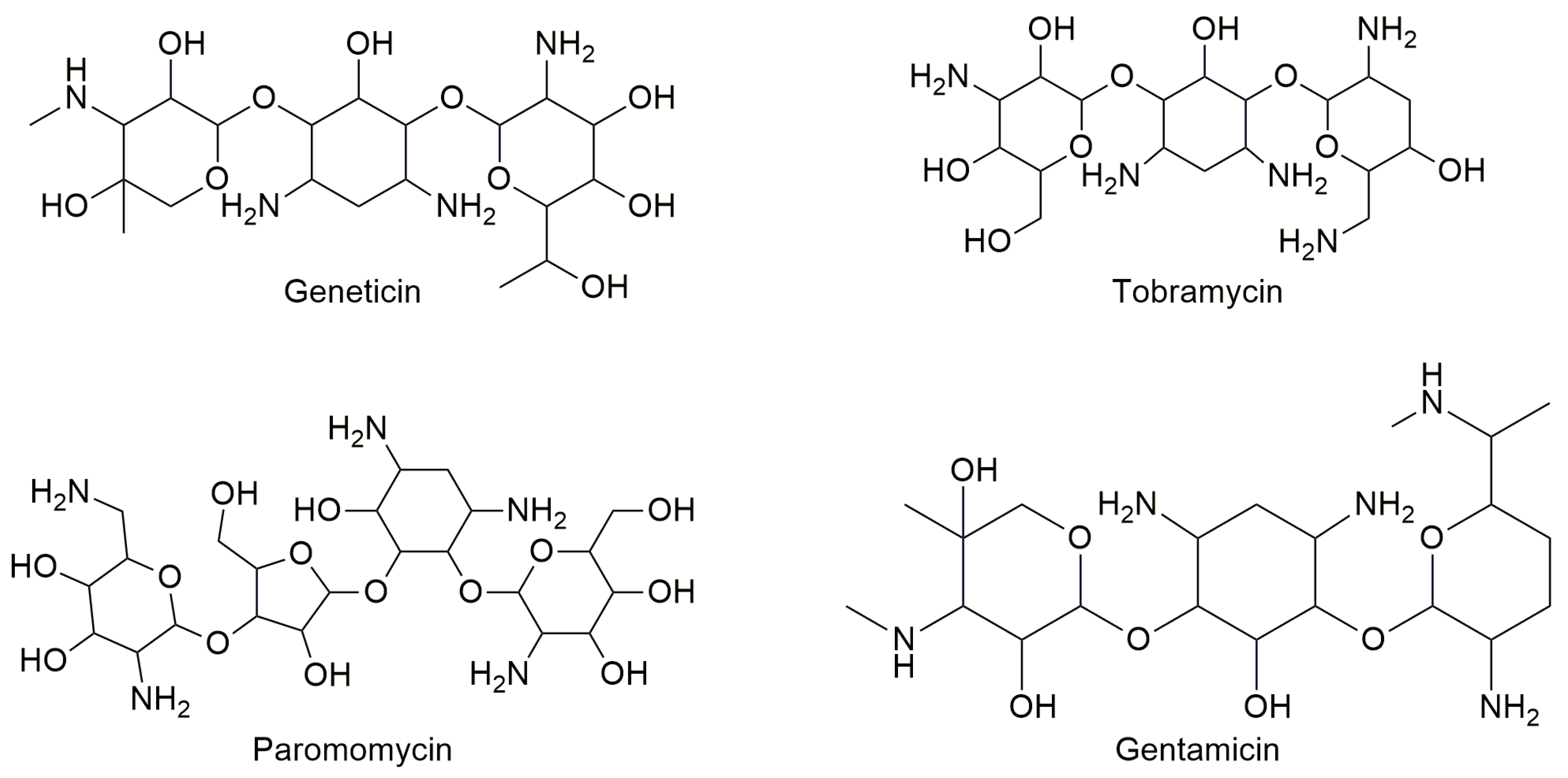
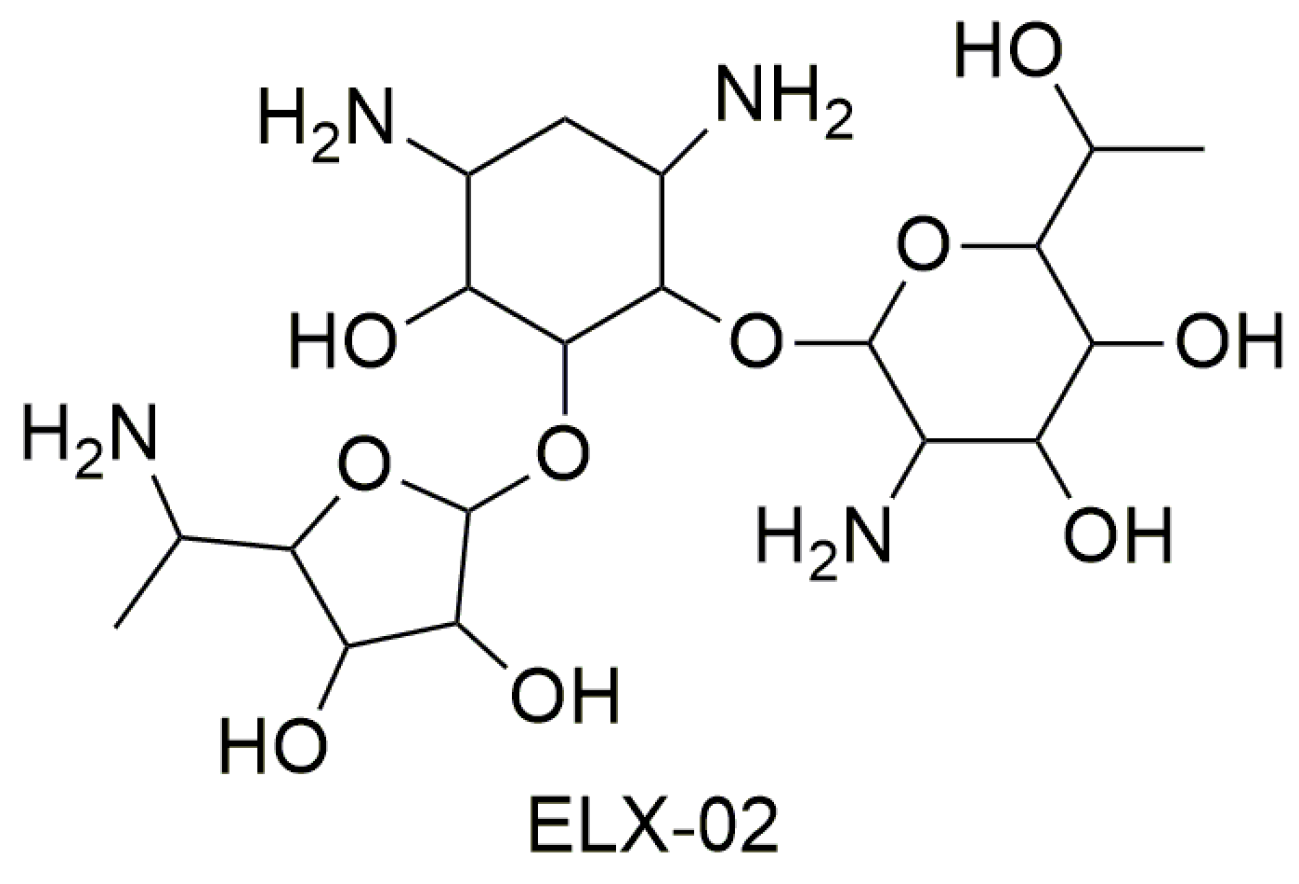
3.1. Aminoglycosides
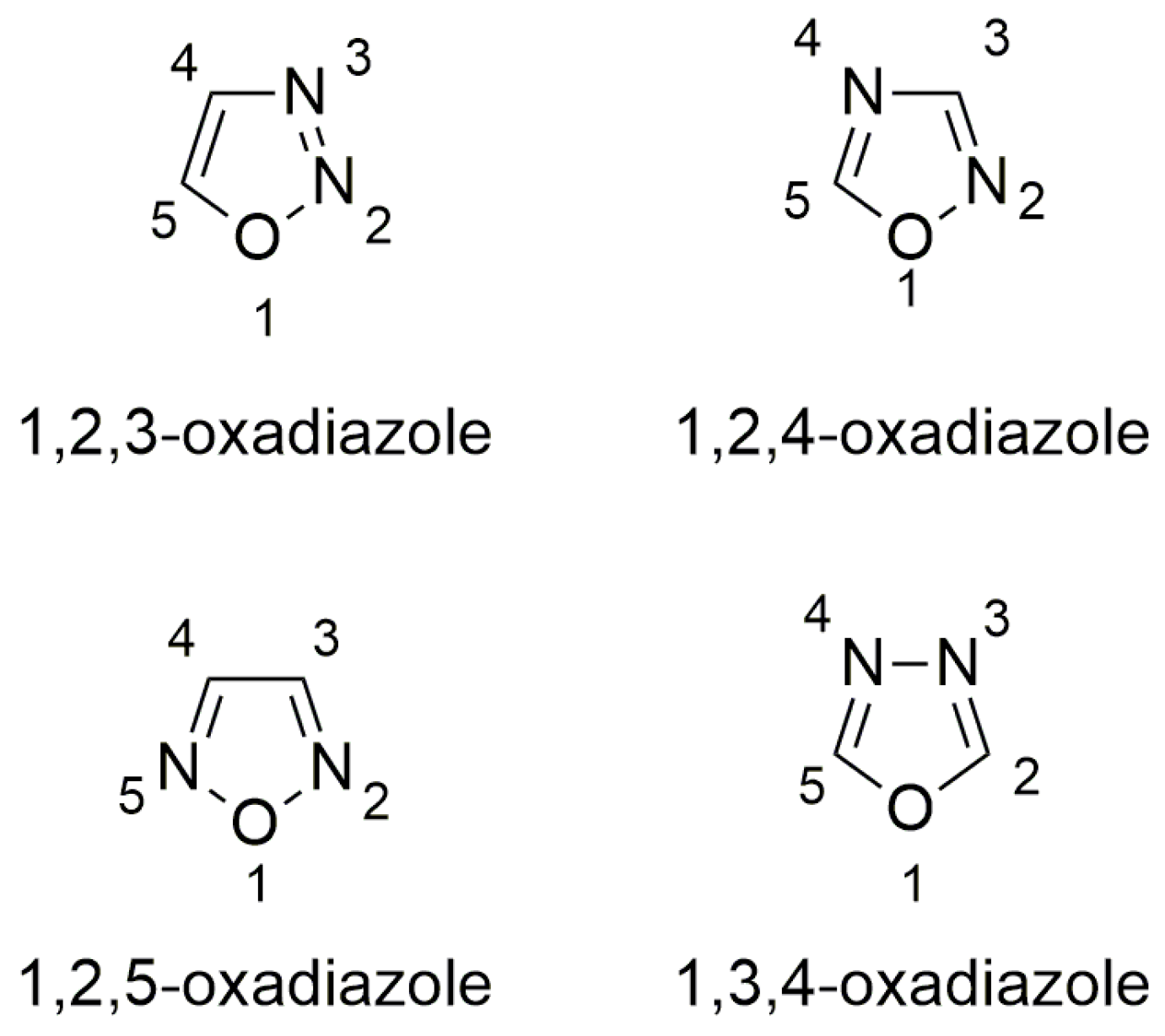
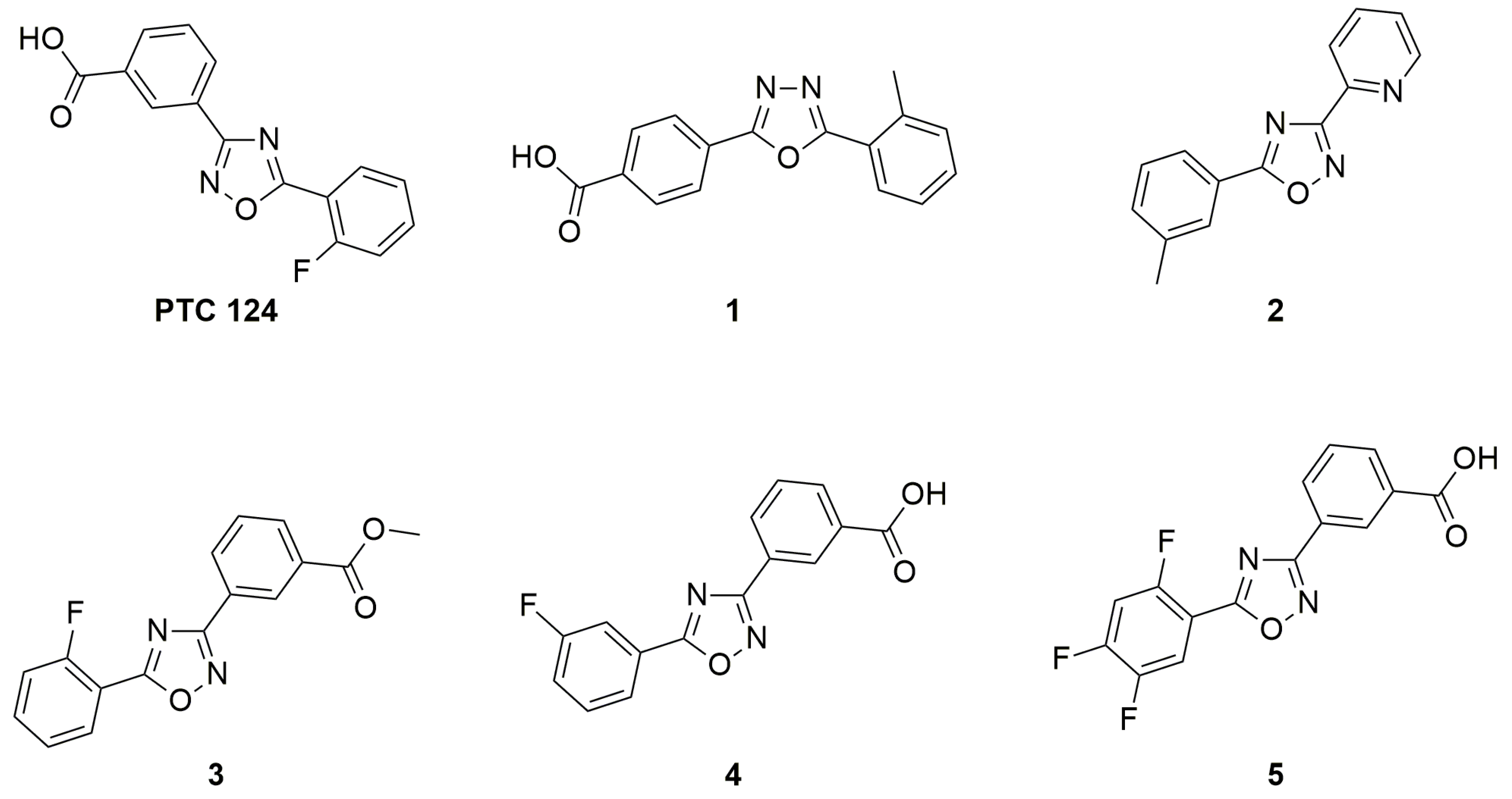
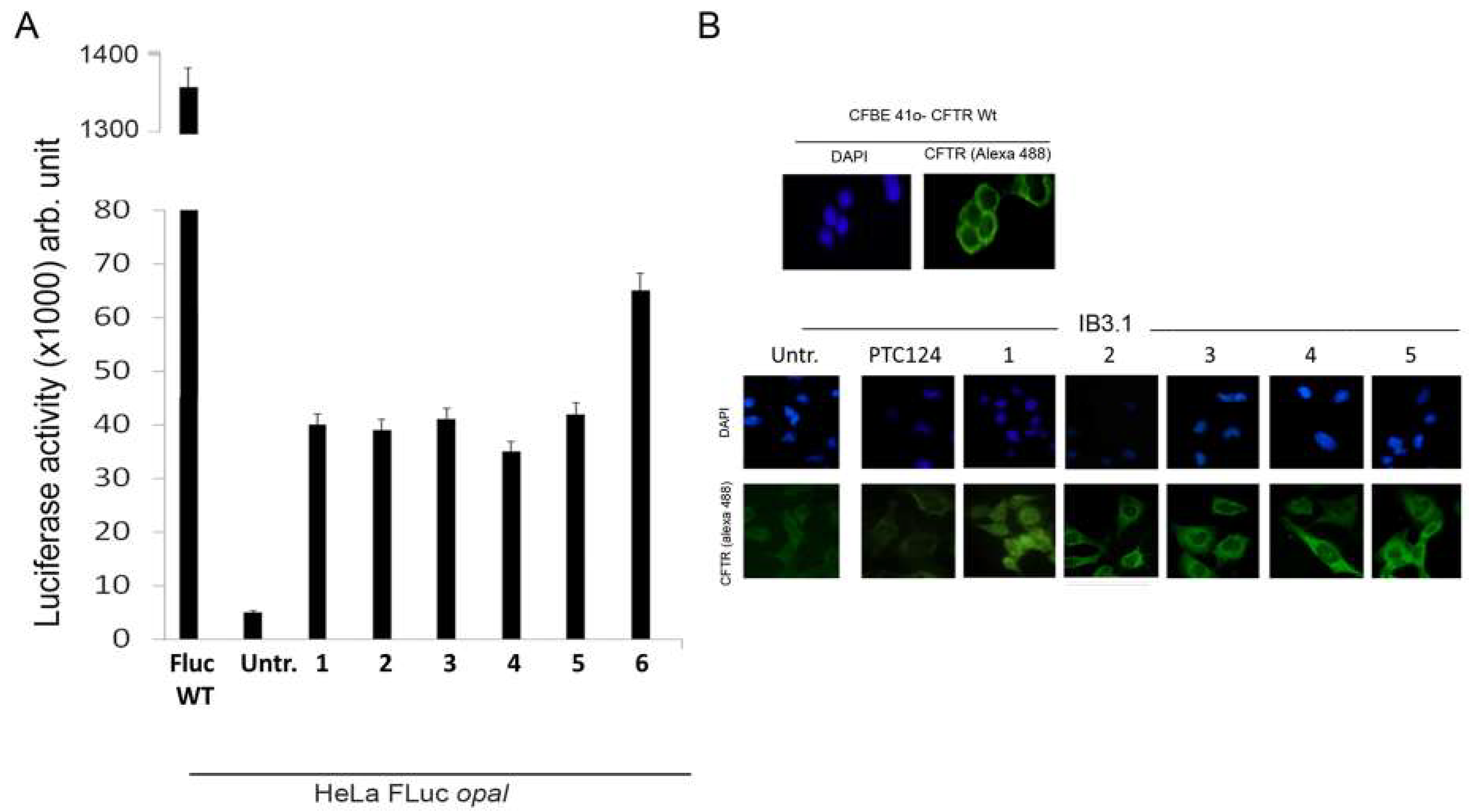
3.2. Oxadiazoles
3.3. Miscellanea
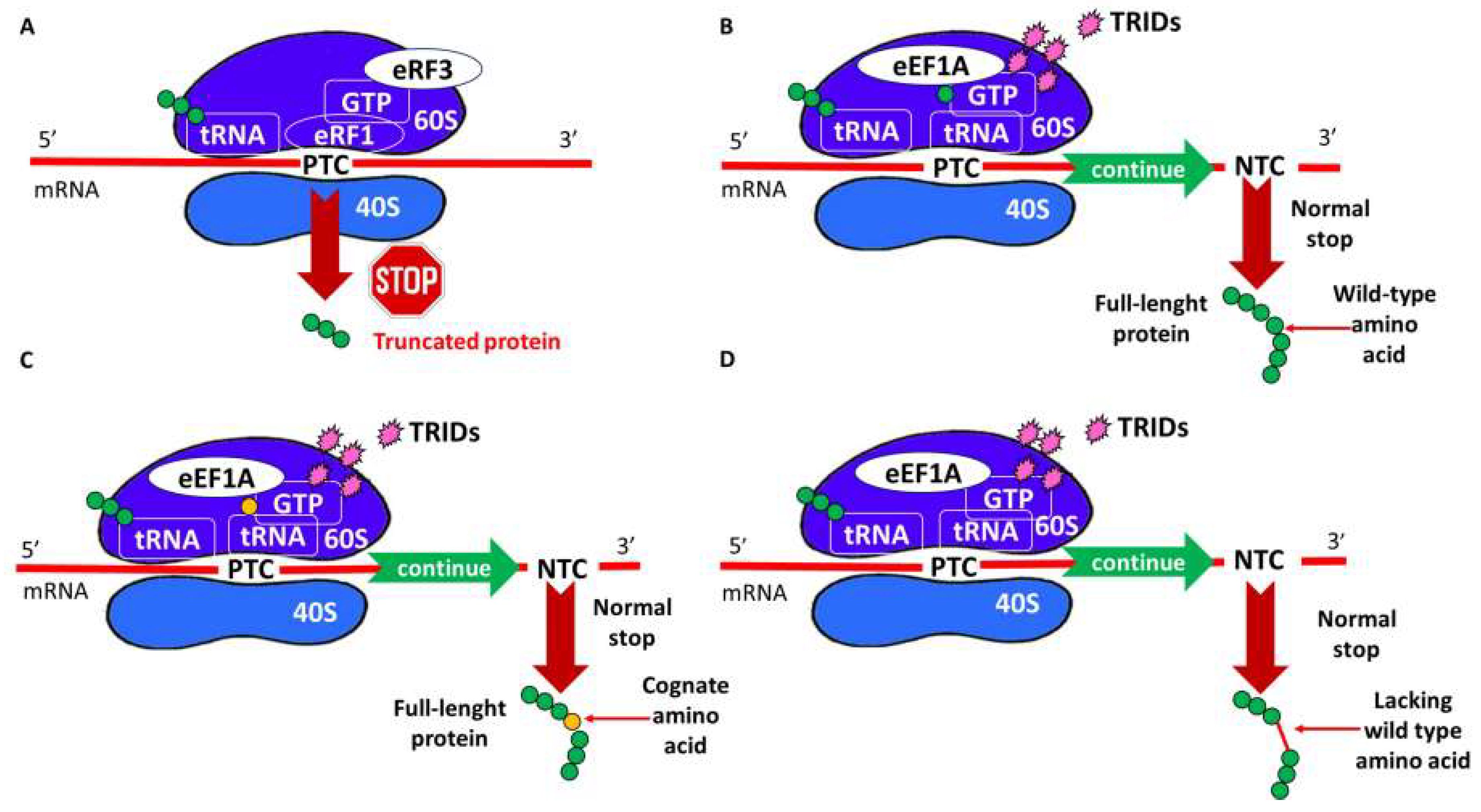
4. TRID Mechanism of Action
5. Increasing TRID Efficiency
5.1. Nonsense-Mediated mRNA Decay (NMD) Inhibition
5.2. Correctors and Potentiators
6. Conclusions and Perspectives
Funding
Conflicts of Interest
Abbreviations
| ACE-tRNAs | Anticodon engineered transfer RNAs |
| AT | Ataxia telangiectasia |
| CF | Cystic fibrosis |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMD | Duchenne muscular dystrophy |
| FTR cells | Fisher rat thyroid cells |
| GFP | Green Fluorescent Protein |
| HS | Hurler’s syndrome |
| IFD | Induced Fit Docking |
| MD | Molecular Dynamics |
| MM-GBSA | molecular mechanics /generalized Born and surface area continuum solvation |
| NMD | Nonsense-mediated mRNA decay |
| NonSups | Nonsense suppressors |
| pFLuc-wt | Plasmid firefly luciferase-wild type |
| PTC | Premature stop codon |
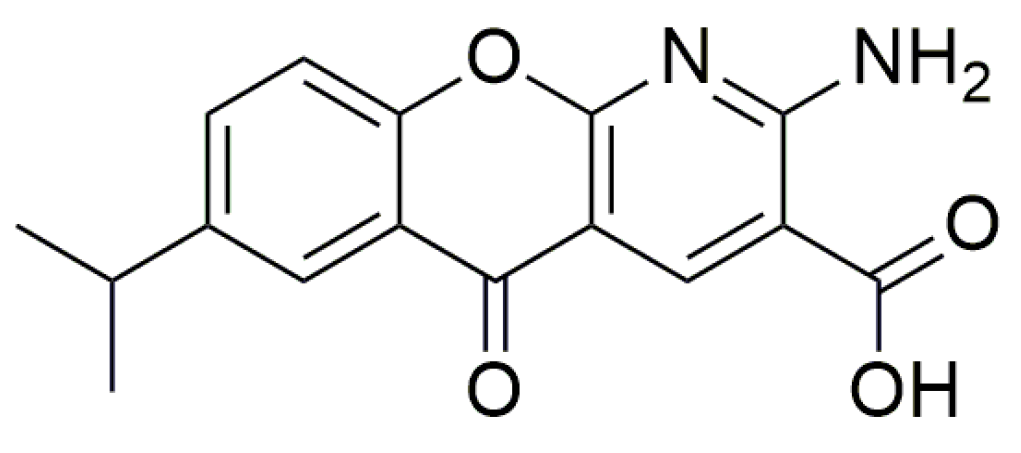
| PTC124 | Ataluren or 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl] benzoic acid |
| QPLD | Quantum Polarized Ligand Docking |
| RF | Release factor |
| RLuc | Renilla reniformis luciferase |
| TRIDs | Translational Readthrough-Inducing Drugs |
| Upf1 | Regulator of nonsense transcripts 1 |
| USH | Usher’s syndrome |
| YFP | Yellow fluorescent protein |
References
- Lakhotia, S.C. Central dogma, selfish DNA and noncoding Rnas: A historical perspective. Proc. Indian Natl. Sci. Acad. 2018, 84, 415–427. [Google Scholar] [CrossRef]
- Crick, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, M.D.; Pfleger, B.F. Transcription control engineering and applications in synthetic biology. Synth. Syst. Biotechnol. 2017, 2, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Litwack, G. Protein Biosynthesis. In Human Biochemistry; Litwack, G., Ed.; Publisher: Los Angeles, CA, USA, 2018; Chapter 11; pp. 319–336. [Google Scholar]
- Antonarakis, S.E.; Cooper, D.N. Human Gene Mutation in Inherited Disease: Molecular Mechanisms and Clinical Consequences. In Emery Rimoin’s Princ Pract Med Genet, 6th ed.; Rimoin, D., Pyeritz, R., Bruce, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Chapter 7; pp. 1–48. [Google Scholar]
- Peltz, S.W.; Morsy, M.; Welch, E.M.; Jacobson, A. Ataluren as an Agent for Therapeutic Nonsense Suppression. Annu. Rev. Med. 2013, 64, 407–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejeune, F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017, 50, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loudon, J.A. Repurposing Amlexanox as a ‘Run the Red Light Cure-All’ with Read-Through—A ‘No-Nonsense’ Approach to Personalised Medicine. J. Bioanal. Biomed. 2013, 5, 79–96. [Google Scholar] [CrossRef]
- Ng, M.Y.; Zhang, H.; Weil, A.; Singh, V.; Jamiolkowski, R.; Baradaran-Heravi, A.; Roberge, M.; Jacobson, A.; Friesen, W.; Welch, E.; et al. New in Vitro Assay Measuring Direct Interaction of Nonsense Suppressors with the Eukaryotic Protein Synthesis Machinery. ACS Med. Chem. Lett. 2018, 9, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Ardiçli, D.; Haliloǧlu, G.; Alikaşifoǧlu, M.; Topaloǧlu, H. Diagnostic Pathway to Nonsense Mutation Dystrophinopathy: A Tertiary-Center, Retrospective Experience. Neuropediatrics 2019, 50, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Sossi, V.; Giuli, A.; Vitali, T.; Tiziano, F.; Mirabella, M.; Antonelli, A.; Neri, G.; Brahe, C. Premature termination mutations in exon 3 of the SMN1 gene are associated with exon skipping and a relatively mild SMA phenotype. Eur. J. Hum. Genet. 2001, 9, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, L.; Sun, X.; Lu, Z.; Suo, X.; Li, J.; Peng, J.; Peng, R. A novel mutation in VRK1 associated with distal spinal muscular atrophy. J. Hum. Genet. 2019, 64, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Turner, A.N.; Chen, M.; Brosius, S.N.; Schoeb, T.R.; Messiaen, L.M.; Bedwell, D.M.; Zinn, K.R.; Anastasaki, C.; Gutmann, D.H.; et al. Mice with missense and nonsense NF1 mutations display divergent phenotypes compared with human neurofibromatosis type I. Dis. Model. Mech. 2016, 9, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.A.; Lee, W.; Cai, C.; Gambin, T.; Nõupuu, K.; Sujirakul, T.; Ayuso, C.; Jhangiani, S.; Muzny, D.; Boerwinkle, E.; et al. New syndrome with retinitis pigmentosa is caused by nonsense mutations in retinol dehydrogenase RDH11. Hum. Mol. Genet. 2014, 23, 5774–5780. [Google Scholar] [CrossRef]
- Kiser, K.; Webb-Jones, K.D.; Bowne, S.J.; Sullivan, S.L.; Daiger, S.P.; Birch, D.G. Time Course of Disease Progression of PRPF31-mediated Retinitis Pigmentosa. Am. J. Ophthalmol. 2019, 200, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Schiff, M.; Tikkanen, R. Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Shen, R.; Zhang, W.; Mao, B.; Shi, Q.; Zhou, R.; Liu, Z.; Zeng, B.; Chen, X.; Zhang, C.; et al. Clinical diagnosis and genetic counseling of atypical ataxia-telangiectasia in a Chinese family. Mol. Med. Rep. 2019, 19, 3441–3448. [Google Scholar] [CrossRef] [PubMed]
- Chernushyn, S.; Gulkovskyi, R.; Livshits, L. Novel Mutation in the MECP2 Gene Identified in a Group of Rett Syndrome Patients from Ukraine. Cytol. Genet. 2018, 52, 294–298. [Google Scholar] [CrossRef]
- Bezzerri, V.; Bardelli, D.; Morini, J.; Vella, A.; Cesaro, S.; Sorio, C.; Biondi, A.; Danesino, C.; Farruggia, P.; Assael, B.M.; et al. Ataluren-driven restoration of Shwachman-Bodian-Diamond syndrome protein function in Shwachman-Diamond syndrome bone marrow cells. Am. J. Hematol. 2017, 93, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Overlack, N.; Wolfrum, U.; Nagel-Wolfrum, K. PTC124-Mediated Translational Readthrough of a Nonsense Mutation Causing Usher Syndrome Type 1C. Hum. Gene Ther. 2011, 22, 537–547. [Google Scholar] [CrossRef]
- James, P.D.; Raut, S.; Rivard, G.E.; Poon, M.C.; Warner, M.; McKenna, S.; Leggo, J.; Lillicrap, D. Aminoglycoside Suppression of Nonsense Mutations in Severe Hemophilia. Blood 2005, 106, 3043–3048. [Google Scholar] [CrossRef]
- Shalev, M.; Baasov, T. When Proteins Start to Make Sense: Fine-tuning Aminoglycosides for PTC Suppression Therapy. Medchemcomm 2014, 5, 1092–1105. [Google Scholar] [CrossRef]
- Bordeira-Carriço, R.; Pêgo, A.P.; Santos, M.; Oliveira, C. Cancer syndromes and therapy by stop-codon readthrough. Trends Mol. Med. 2012, 18, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Colemeadow, J.; Joyce, H.; Turcanu, V. Precise treatment of cystic fibrosis–current treatments and perspectives for using CRISPR. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 169–180. [Google Scholar] [CrossRef]
- Harrison, P.T.; Hoppe, N.; Martin, U. Gene editing & stem cells. J. Cyst. Fibros. 2018, 17, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Montagna, C.; Petris, G.; Casini, A.; Maule, G.; Franceschini, G.M.; Zanella, I.; Conti, L.; Arnoldi, F.; Burrone, O.R.; Zentilin, L.; et al. VSV-G-Enveloped Vesicles for Traceless Delivery of CRISPR-Cas9. Mol. Ther. Nucleic Acids 2018, 12, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Pibiri, I.; Lentini, L.; Melfi, R.; Gallucci, G.; Pace, A.; Spinello, A.; Barone, G.; Di Leonardo, A. Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur. J. Med. Chem. 2015, 101, 236–244. [Google Scholar] [CrossRef]
- Midgley, J. A breakthrough in readthrough? Could geneticin lead the way to effective treatment for cystinosis nonsense mutations? Pediatr. Nephrol. 2019. [CrossRef]
- Nagel-Wolfrum, K.; Möller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting Nonsense Mutations in Diseases with Translational Read-Through-Inducing Drugs (TRIDs). BioDrugs 2016, 30, 49–74. [Google Scholar] [CrossRef]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, T.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, I.; Lentini, L.; Tutone, M.; Melfi, R.; Pace, A.; Di Leonardo, A. Exploring the readthrough of nonsense mutations by non-acidic Ataluren analogues selected by ligand-based virtual screening. Eur. J. Med. Chem. 2016, 122, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Friesen, W.J.; Johnson, B.; Sierra, J.; Zhuo, J.; Vazirani, P.; Xue, X.; Tomizawa, Y.; Baiazitov, R.; Morrill, C.; Ren, H.; et al. The minor gentamicin complex component, X2, is a potent premature stop codon readthrough molecule with therapeutic potential. PLoS ONE 2018, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Leubitz, A.; Frydman-Marom, A.; Sharpe, N.; van Duzer, J.; Campbell, K.C.M.; Vanhoutte, F. Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of ELX-02, a Potential Treatment for Genetic Disorders Caused by Nonsense Mutations, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bidou, L.; Bugaud, O.; Belakhov, V.; Baasov, T.; Namy, O. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 2017, 14, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karijolich, J.; Yu, Y.T. Therapeutic suppression of premature termination codons: Mechanisms and clinical considerations (Review). Int. J. Mol. Med. 2014, 34, 355–362. [Google Scholar] [CrossRef]
- Floquet, C.; Hatin, I.; Rousset, J.P.; Bidou, L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PLoS Genet. 2012, 8, e1002608. [Google Scholar] [CrossRef]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic Aminoglycosides Efficiently Suppress Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations and Are Enhanced by Ivacaftor. Am. J. Respir. Cell Mol. Biol. 2014, 50, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Fraser, W. 1, 2, 3-Oxadiazoles. In Comprehensive Heterocyclic Chemistry III, 1st ed.; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 5, p. 211. [Google Scholar]
- Pace, A.; Buscemi, S.; Palumbo Piccionello, A.; Pibiri, I. Recent Advances in the Chemistry of 1, 2, 4-Oxadiazoles. In Advances Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 116, pp. 85–136. [Google Scholar]
- Pace, A.; Pierro, P. The new era of 1, 2, 4-oxadiazoles. Org. Biomol. Chem. 2009, 7, 4337–4348. [Google Scholar] [CrossRef]
- Paton, R.M. 1, 2, 5-Oxadiazoles. In Comprehensive Heterocyclic Chemistry II, 1st ed.; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 4, pp. 229–265. [Google Scholar]
- Salahuddin, A.; Mazumder, M.; Yar, M.S.; Mazumder, R.; Chakraborthy, G.S.; Ahsan, M.J.; Rahman, M.U. Updates on synthesis and biological activities of 1, 3, 4-oxadiazole: A review. Synth. Commun. 2017, 47, 1805–1847. [Google Scholar] [CrossRef]
- Paun, A.; Hadade, N.D.; Paraschivescu, C.C.; Matache, M. 1, 3, 4-Oxadiazoles as luminescent materials for organic light emitting diodes via cross-coupling reactions. J. Mater. Chem. C 2016, 4, 8596–8610. [Google Scholar] [CrossRef]
- Palumbo Piccionello, A.; Pace, A.; Buscemi, S. Rearrangements of 1, 2, 4-Oxadiazole: “One Ring to Rule Them All”. Chem. Heterocycl. Compd. 2017, 53, 936–947. [Google Scholar] [CrossRef]
- Palumbo, F.S.; Di Stefano, M.; Palumbo Piccionello, A.; Fiorica, C.; Pitarresi, G.; Pibiri, I.; Buscemi, S.; Giammona, G. Perfluorocarbon functionalized hyaluronic acid derivatives as oxygenating systems for cell culture. RSC Adv. 2014, 4, 22894–22901. [Google Scholar] [CrossRef]
- Pibiri, I.; Pace, A.; Buscemi, S.; Causin, V.; Rastrelli, F.; Saielli, G. Oxadiazolyl-pyridines and perfluoroalkyl-carboxylic acids as building blocks for protic ionic liquids: Crossing the thin line between ionic and hydrogen bonded materials. Phys. Chem. Chem. Phys. 2012, 14, 14306–14314. [Google Scholar] [CrossRef] [PubMed]
- Palumbo Piccionello, A.; Calabrese, A.; Pibiri, I.; Giacalone, V.; Pace, A.; Buscemi, S. Synthesis of Fluorinated Bent-Core Mesogens (BCMs) Containing the 1, 2, 4-Oxadiazole Ring. J. Heterocycl. Chem. 2016, 53, 1935–1940. [Google Scholar] [CrossRef]
- Palumbo Piccionello, A.; Guarcello, A.; Calabrese, A.; Pibiri, I.; Pace, A.; Buscemi, S. Synthesis of fluorinated oxadiazoles with gelation and oxygen storage ability. Org. Biomol. Chem. 2012, 10, 3044–3052. [Google Scholar] [CrossRef]
- Pibiri, I.; Beneduci, A.; Carraro, M.; Causin, V.; Casella, G.; Corrente, G.A.; Chidichimo, G.; Pace, A.; Riccobono, A.; Saielli, G. Mesomorphic and electrooptical properties of viologens based on non-symmetric alkyl/polyfluoroalkyl functionalization and on an oxadiazolyl-extended bent core. J. Mater. Chem. C 2019. [Google Scholar] [CrossRef]
- Huang, X.C.; Wang, Z.J.; Guo, T.; Qin, M.N.; Liu, M.; Qiu, S.J. Review on Energetic Compounds Based on 1, 2, 4-Oxadiazoles. Hanneng Cailiao/Chin. J. Energetic 2017, 25, 603–611. [Google Scholar] [CrossRef]
- Fouad, F.S.; Ness, T.; Wang, K.; Ruth, C.E.; Britton, S.; Twieg, R.J. Biphenylyl-1, 2, 4-oxadiazole based liquid crystals–synthesis, mesomorphism, effect of lateral monofluorination. Liq. Cryst. 2019. [Google Scholar] [CrossRef]
- Palumbo Piccionello, A.; Musumeci, R.; Cocuzza, C.; Fortuna, C.G.; Guarcello, A.; Pierro, P.; Pace, A. Synthesis and preliminary antibacterial evaluation of Linezolid-like 1, 2, 4-oxadiazole derivatives. Eur. J. Med. Chem. 2012, 50, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Tarasenko, M.; Duderin, N.; Sharonova, T.; Baykov, S.; Shetnev, A.; Smirnov, A.V. Room-temperature synthesis of pharmaceutically important carboxylic acids bearing the 1, 2, 4-oxadiazole moiety. Tetrahedron Lett. 2017, 58, 3672–3677. [Google Scholar] [CrossRef]
- Tarasenko, M.; Sidneva, V.; Belova, A.; Romanycheva, A.; Sharonova, T.; Baykov, S.; Shetnev, A.; Kofanov, E.; Kuznetsov, M.A. An efficient synthesis and antimicrobial evaluation of 5-alkenyl-and 5-styryl-1, 2, 4-oxadiazoles. Arkivoc 2018, 458–470. [Google Scholar] [CrossRef]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in Medicinal Chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Rottini, E.; Marri, G.; Calandra, P. The recent introduction in therapy of a new antitussive drug: Oxolamine. Minerva Med. 1961, 52, 3758–3760. [Google Scholar]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Paz, O.G.; Hazuda, D.J.; et al. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef]
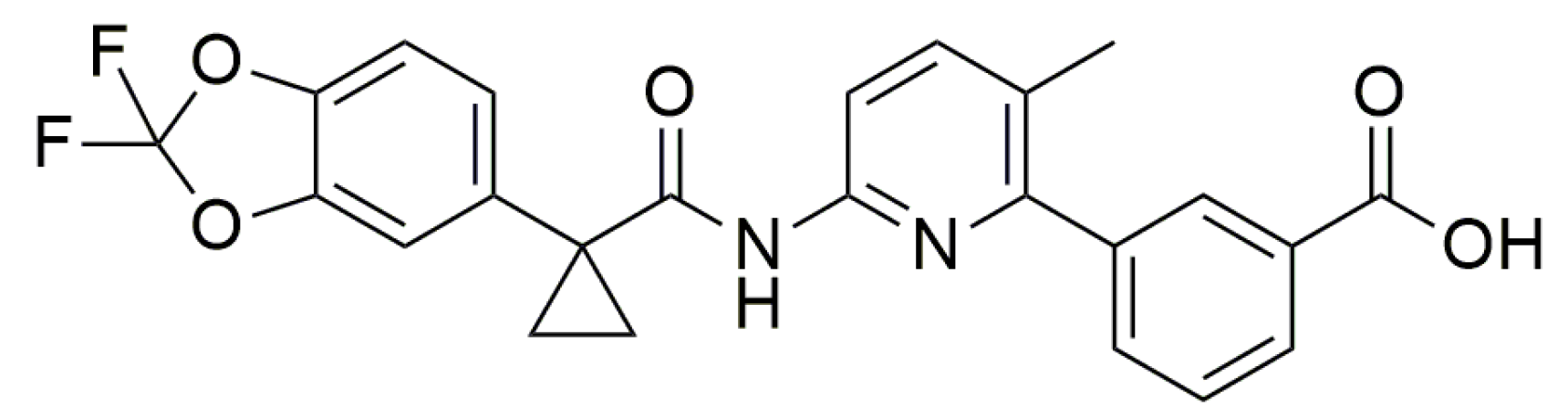
- James, N.D.; Growcott, J.W. Zibotentan. Drugs Fut. 2009, 34, 624. [Google Scholar] [CrossRef]
- Hirawat, S.; Welch, E.M.; Elfring, G.L.; Northcutt, V.J.; Paushkin, S.; Hwang, S.; Leonard, E.M.; Almstead, N.G.; Ju, W.; Peltz, S.W.; et al. Safety, Tolerability, and Pharmacokinetics of PTC124, a Nonaminoglycoside Nonsense Mutation Suppressor, Following Single- and Multiple-Dose Administration to Healthy Male and Female Adult Volunteers. J. Clin. Pharmacol. 2007, 47, 430–444. [Google Scholar] [CrossRef] [Green Version]
- Haas, M.; Vlcek, V.; Balabanov, P.; Salmonson, T.; Bakchine, S.; Markey, G.; Weise, M.; Schlosser-Weber, G.; Brohmann, H.; Yerro, C.P.; et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul. Disord. 2015, 25, 5–13. [Google Scholar] [CrossRef]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef]
- Siddiqui, N.; Sonenberg, N. Proposing a mechanism of action for ataluren. Proc. Natl. Acad. Sci. USA 2016, 113, 12353–12355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auld, D.S.; Thorne, N.; Maguire, W.F.; Inglese, J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. USA 2009, 106, 3585–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auld, D.S.; Lovell, S.; Thorne, N.; Lea, W.A.; Maloney, D.J.; Shen, M.; Rai, G.; Battaile, K.P.; Thomas, C.J.; Simeonov, A.; et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl. Acad. Sci. USA 2010, 107, 4878–4883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, B.; Leszyk, J.B.; Mangus, D.A.; Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3038–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef]
- Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; Palumbo Piccionello, A.; Pibiri, I. Toward a Rationale for the PTC124 (Ataluren) Promoted Readthrough of Premature Stop Codons: A Computational Approach and GFP-Reporter Cell-Based Assay. Mol. Pharm. 2014, 11, 653–664. [Google Scholar] [CrossRef]
- Gitter, A.H.; Schulzke, J.D.; Sorgenfrei, D.; Fromm, M. Ussing chamber for high-frequency transmural impedance analysis of epithelial tissues. J. Biochem. Biophys. Methods 1997, 35, 81–88. [Google Scholar] [CrossRef]
- Baumann, C.T.; Lim, C.S.; Hager, G.L. Simultaneous Visualization of the Yellow and Green Forms of the Green Fluorescent Protein in Living Cells. J. Histochem. Cytochem. 1998, 46, 1073–1076. [Google Scholar] [CrossRef]
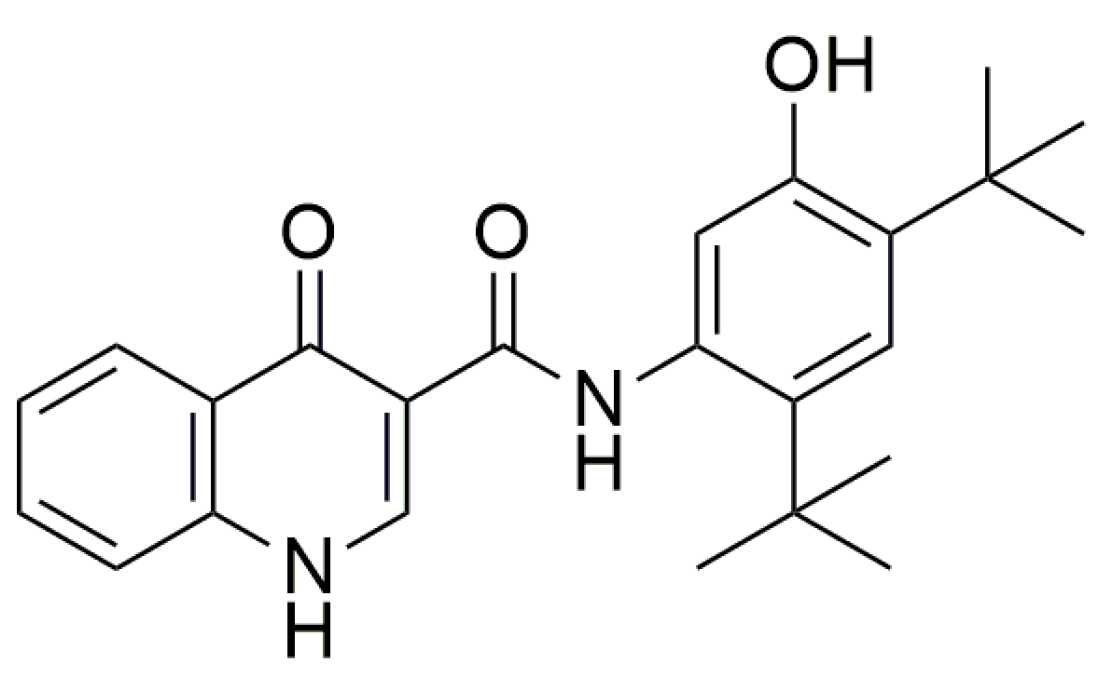
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Déprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 1–14. [Google Scholar] [CrossRef]
- Saijo, T.; Kuriki, H.; Ashida, Y.; Makino, H.; Maki, Y. Mechanism of the action of Amoxanox (AA-673) an Orally Active Antiallergic Agent. Int. Archs Allergy Appl. Immun. 1985, 78, 43–50. [Google Scholar] [CrossRef]
- Meng, W.; Dong, Y.; Liu, J.; Wang, Z.; Zhong, X.; Chen, R.; Zhou, H.; Lin, M.; Jiang, L.; Gao, F.; et al. A clinical evaluation of amlexanox oral adhesive pellicles in the treatment of recurrent aphthous stomatitis and comparison with amlexanox oral tablets: A randomized, placebo controlled, blinded, multicenter clinical trial. Trials 2009, 10. [Google Scholar] [CrossRef] [PubMed]
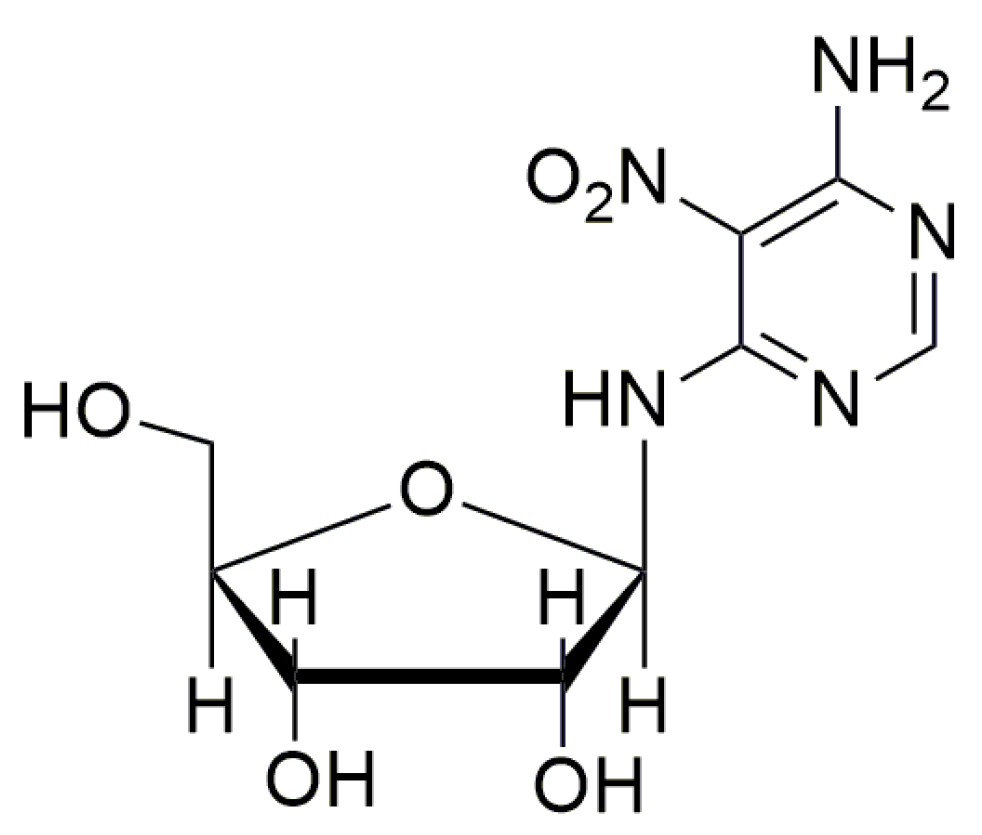
- Friesen, W.J.; Trotta, C.R.; Tomizawa, Y.; Zhuoa, J.; Johnson, B.; Sierra, J.; Roy, B.; Weetall, M.; Hedricka, J.; Sheedy, J.; et al. The nucleoside analog clitocine is a potent and efficacious readthrough agent. RNA 2017, 23, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lueck, J.D.; Yoon, J.S.; Perales-Puchalt, A.; Mackey, A.L.; Infield, D.T.; Behlke, M.A.; Pope, M.R.; Weiner, D.B.; Skach, W.R.; McCray, P.B., Jr.; et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Demeshkina, N.; Jenner, L.; Westhof, E.; Yusupov, M.; Yusupova, G. A new understanding of the decoding principle on the ribosome. Nature 2012, 484, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Shalev, M.; Kondo, J.; Kopelyanskiy, D.; Jaffe, C.L.; Adir, N.; Baasov, T. Identification of the molecular attributes required for aminoglycoside activity against Leishmania. Proc. Natl. Acad. Sci. USA 2013, 110, 13333–13338. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.; Wang, D.; Dai, Y.; Murugesan, S.; Chenna, B.; Clark, J.; Belakhov, V.; Kandasamy, J.; Velu, S.; Baasov, T.; et al. Attenuation of Nonsense-Mediated mRNA Decay Enhances In Vivo Nonsense Suppression. PLoS ONE 2013, 8, e60478. [Google Scholar] [CrossRef] [PubMed]
- Gambari, R.; Breveglieri, G.; Salvatori, F.; Finotti, A.; Borgatti, M. Therapy for Cystic Fibrosis Caused by Nonsense Mutations. In Cystic Fibrosis in the Light of New Research; Wat, D., Ed.; Intech Open: London, UK, 2015; Chapter 13; pp. 309–326. [Google Scholar]
- Keeling, K.M. Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases. Diseases 2016, 4. [Google Scholar] [CrossRef]
- Linde, L.; Kerem, B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008, 24, 552–563. [Google Scholar] [CrossRef]
- Lentini, L.; Melfi, R.; Cancemi, P.; Pibiri, I.; Di Leonardo, A. Caffeine boosts Ataluren’s readthrough activity. Heliyon 2019, 5, e01963. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campofelice, A.; Lentini, L.; Di Leonardo, A.; Melfi, R.; Tutone, M.; Pace, A.; Pibiri, I. Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). Int. J. Mol. Sci. 2019, 20, 3329. https://doi.org/10.3390/ijms20133329
Campofelice A, Lentini L, Di Leonardo A, Melfi R, Tutone M, Pace A, Pibiri I. Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). International Journal of Molecular Sciences. 2019; 20(13):3329. https://doi.org/10.3390/ijms20133329
Chicago/Turabian StyleCampofelice, Ambra, Laura Lentini, Aldo Di Leonardo, Raffaella Melfi, Marco Tutone, Andrea Pace, and Ivana Pibiri. 2019. "Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs)" International Journal of Molecular Sciences 20, no. 13: 3329. https://doi.org/10.3390/ijms20133329
APA StyleCampofelice, A., Lentini, L., Di Leonardo, A., Melfi, R., Tutone, M., Pace, A., & Pibiri, I. (2019). Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). International Journal of Molecular Sciences, 20(13), 3329. https://doi.org/10.3390/ijms20133329