Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease








Abstract
:1. Introduction
2. Results
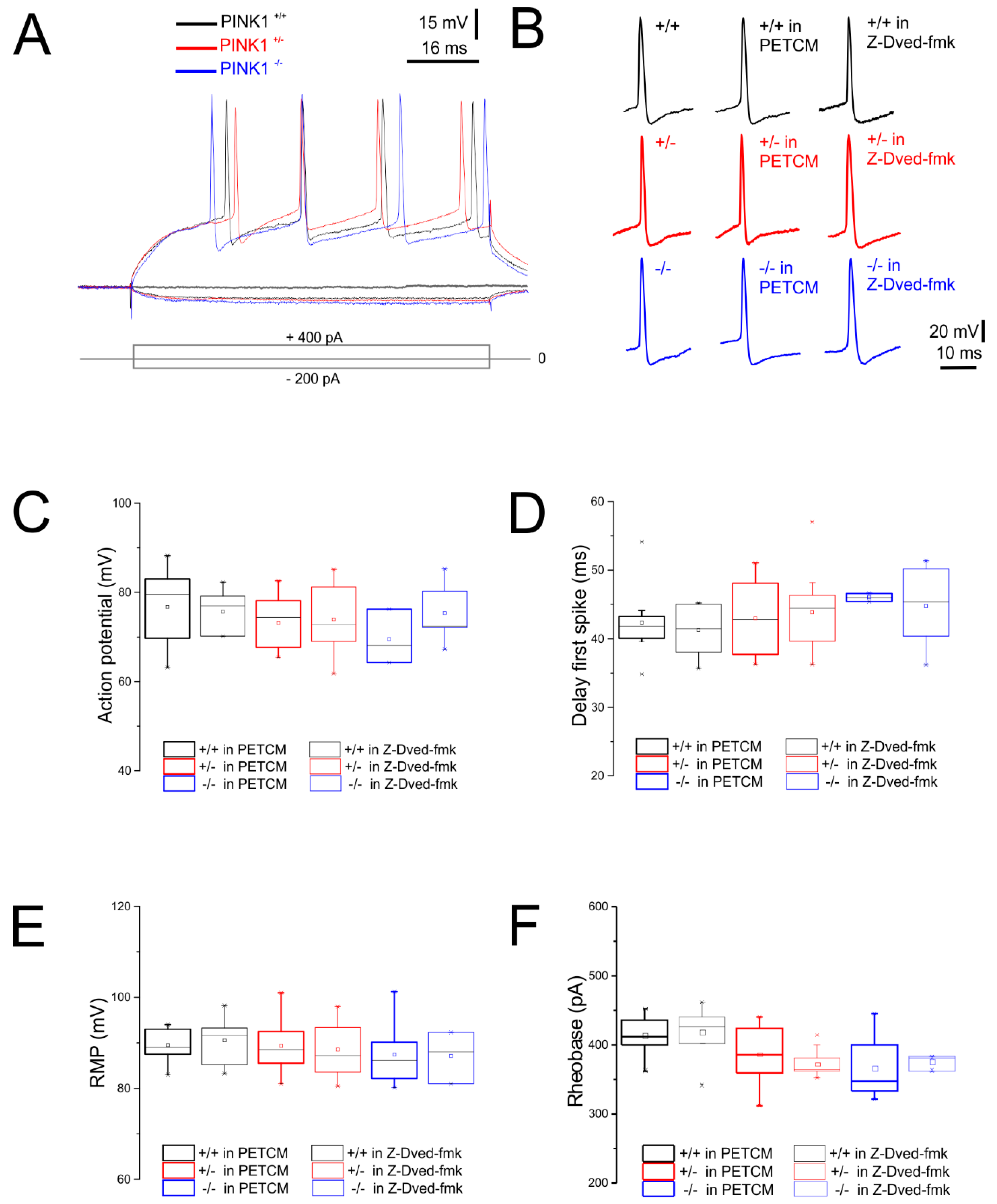
2.1. Normal Intrinsic Membrane Properties of Striatal Medium Spiny Neurons (MSNs) after Pharmacological Modulation of Caspase-3
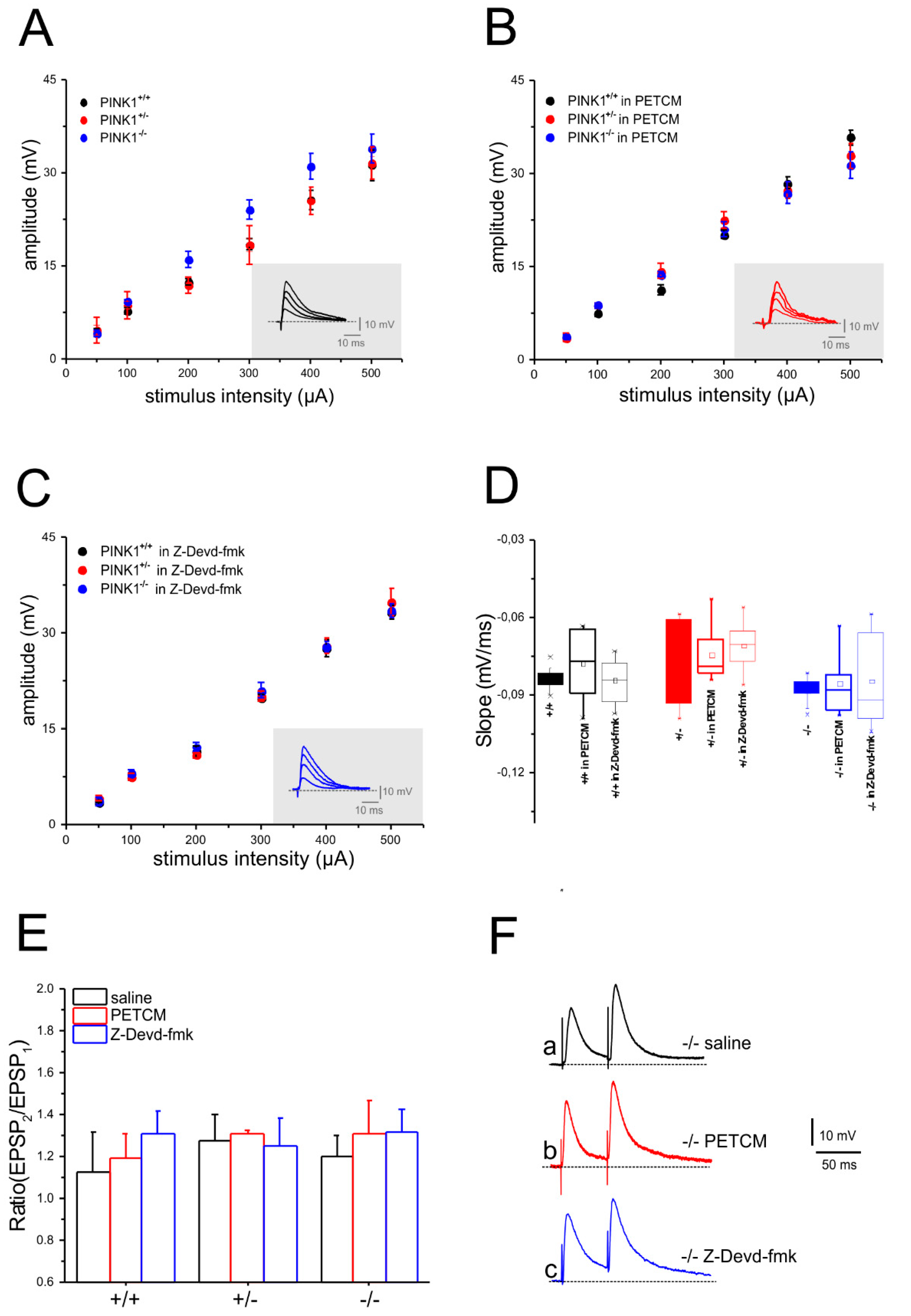
2.2. Unaltered Glutamatergic Short-Term Synaptic Plasticity after Pharmacological Modulation of Caspase-3
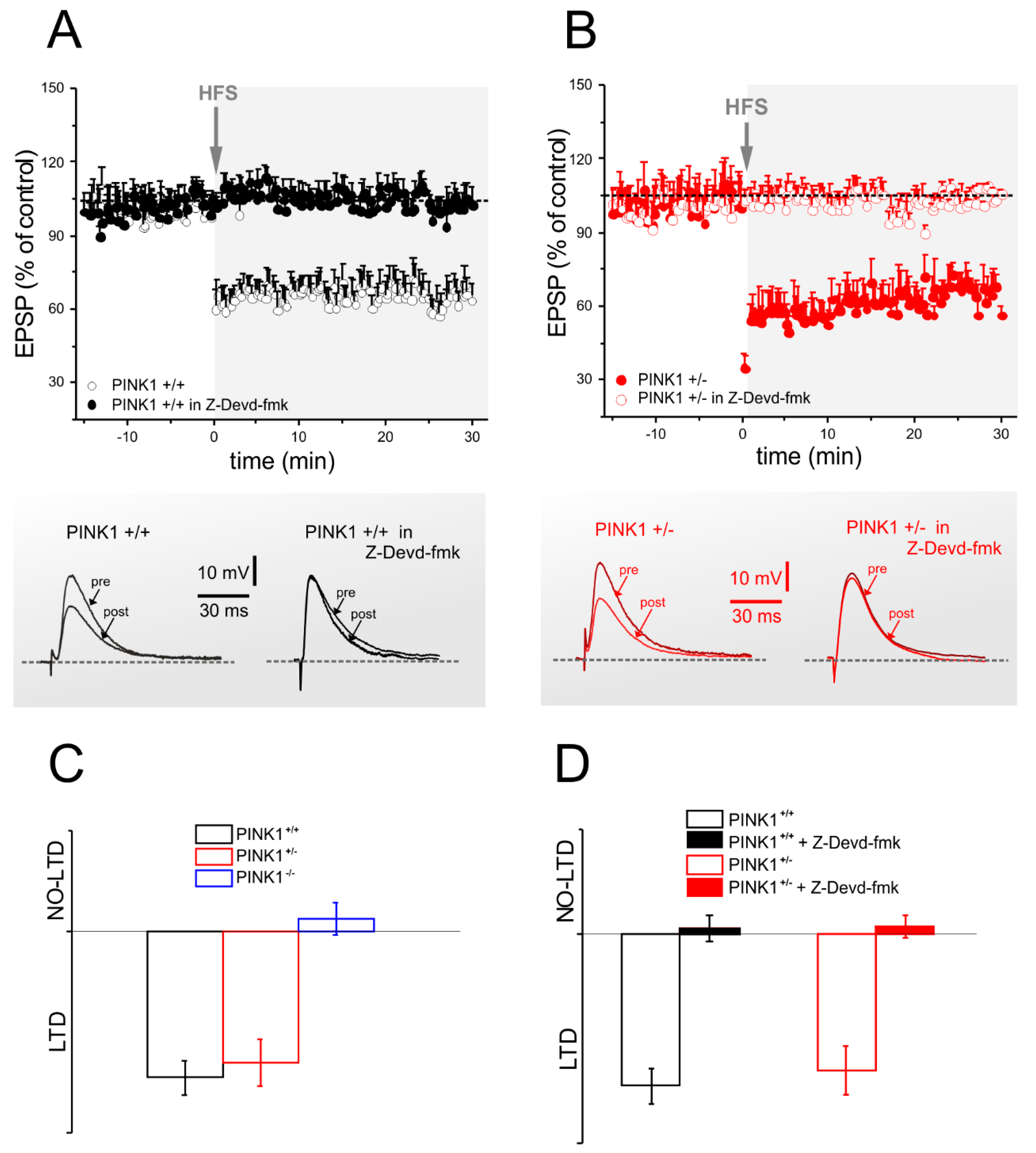
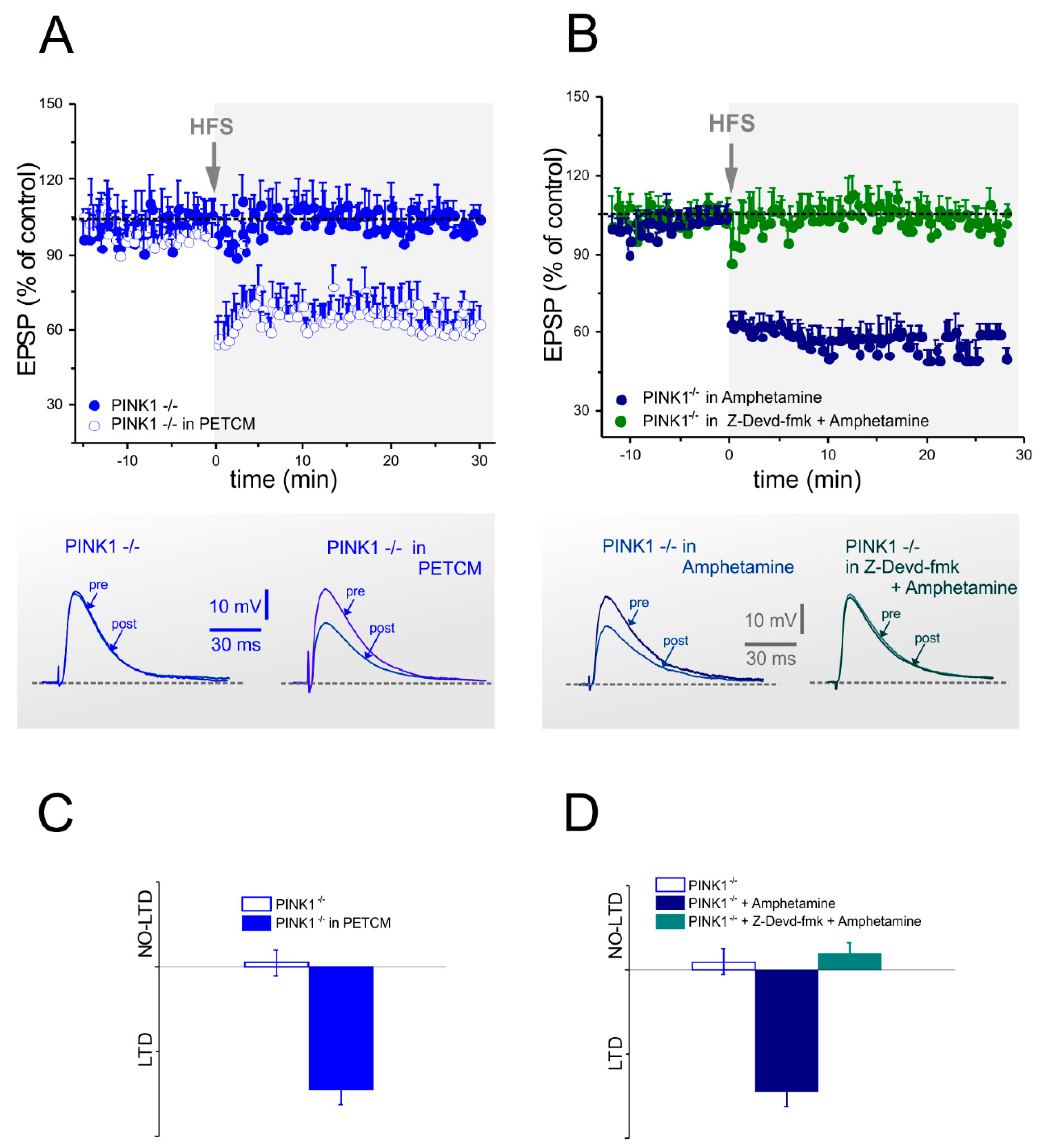
2.3. Corticostriatal LTD Expression Requires Caspase-3 Activation
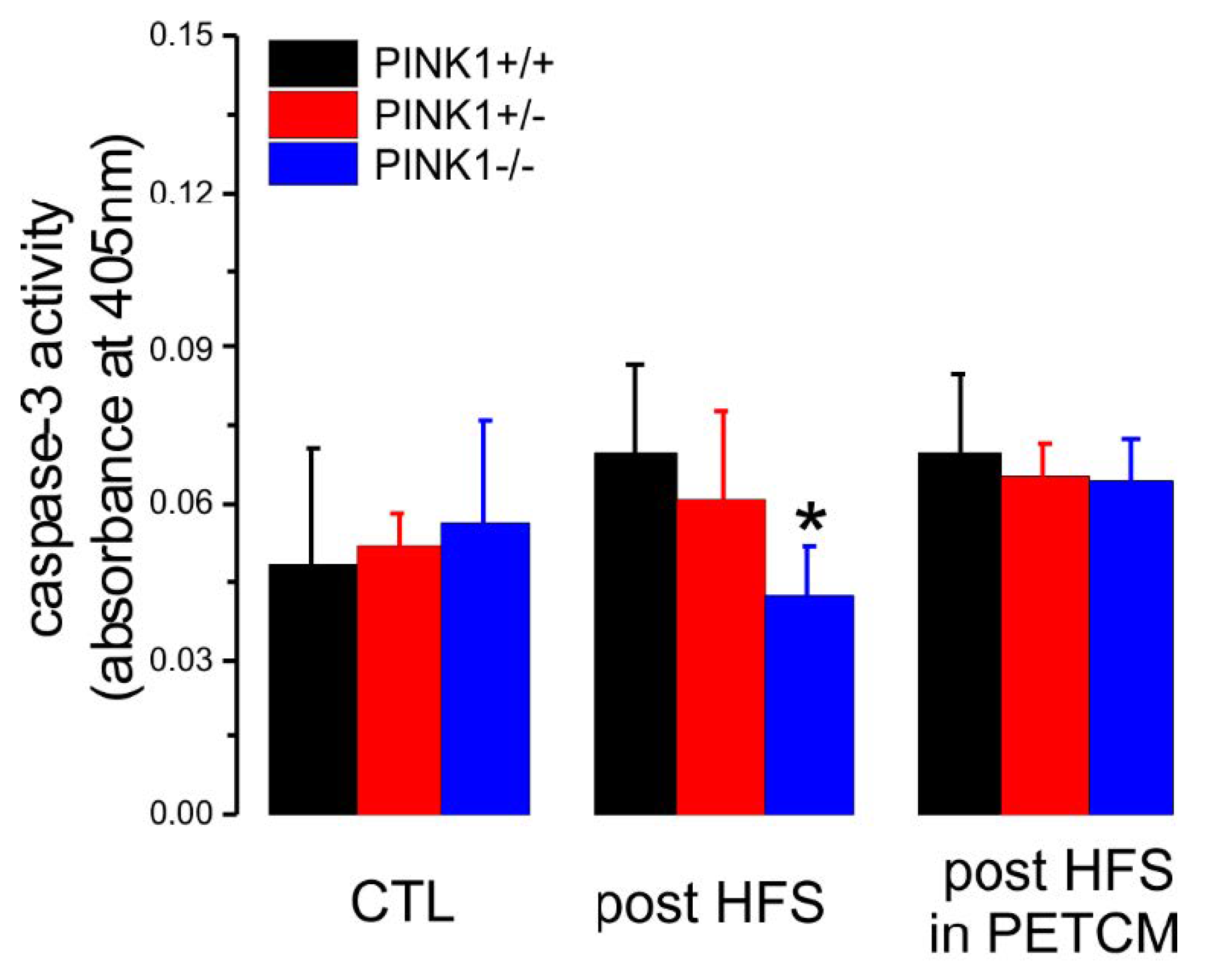
2.4. Caspase-3 Activity is Lower in PINK1−/− Mice after High Frequency Stimulation
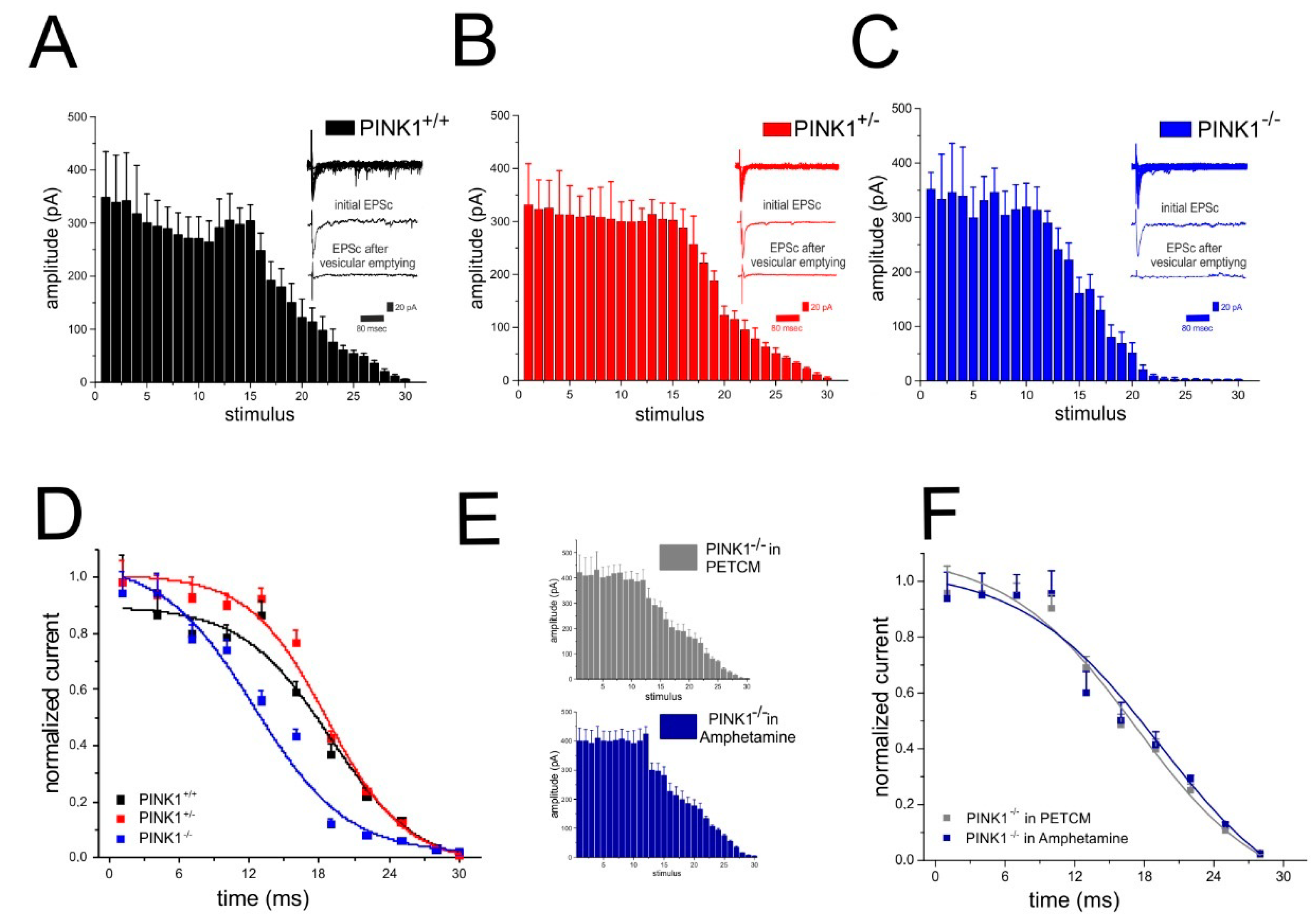
2.5. Analysis of Glutamate Vesicular Release in the Different PINK1 Genotypes
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Electrophysiology
4.3. Caspase-3 Biochemical Assay
4.4. Statistical Analysis
4.5. Drug Sources
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Gilman, C.P.; Mattson, M.P. Do apoptotic mechanisms regulate synaptic plasticity and growth-cone motility? Neuromol. Med. 2002, 2, 197–214. [Google Scholar] [CrossRef]
- Campbell, D.S.; Holt, C.E. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 2003, 37, 939–952. [Google Scholar] [CrossRef]
- Kuo, C.T.; Zhu, S.; Younger, S.; Jan, L.Y.; Jan, Y.N. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 2006, 51, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Kondo, S.; Krzyzanowska, A.; Hiromi, Y.; Truman, J.W. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 2006, 9, 1234–1236. [Google Scholar] [CrossRef]
- Chan, S.L.; Mattson, M.P. Caspase and calpain substrates: Roles in synaptic plasticity and cell death. J. Neurosci. Res. 1999, 58, 167–190. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef]
- Li, Z.; Jo, J.; Jia, J.M.; Lo, S.C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–871. [Google Scholar] [CrossRef]
- Truman, J.W. Metamorphosis of the central nervous system of Drosophila. J. Neurobiol. 1990, 21, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; O’Leary, D.D.M. Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 2005, 28, 127–156. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xiong, W.C.; Mei, L. Caspase-3, shears for synapse pruning. Dev. Cell 2014, 28, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Zheng, T.S.; Na, S.; Kuan, C.; Yang, D.; Karasuyama, H.; Rakic, P.; Flavell, R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 1996, 384, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, S.; Smith, E.D.; Prieto, G.A.; Cotman, C.W. Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci. Bull. 2012, 28, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Blum, S.; Moore, A.N. Caspase activity plays an essential role in long-term memory. Neuroreport 2000, 11, 2811–2816. [Google Scholar] [CrossRef] [PubMed]
- Glazner, G.W.; Chan, S.L.; Lu, C.; Mattson, M.P. Caspase-mediated degradation of AMPA receptor subunits: A mechanism for preventing excitotoxic necrosis and ensuring apoptosis. J. Neurosci. 2000, 20, 3641–3649. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Fu, W.; Salvesen, G.S.; Mattson, M.P. Direct cleavage of AMPA receptor subunit GluR1 and suppression of AMPA currents by caspase-3: Implications for synaptic plasticity and excitotoxic neuronal death. Neuromol. Med. 2002, 1, 69–79. [Google Scholar]
- Ye, B.; Sugo, N.; Hurn, P.D.; Huganir, R.L. Physiological and pathological caspase cleavage of the neuronal RasGEF GRASP-1 as detected using a cleavage site-specific antibody. Neuroscience 2002, 114, 217–227. [Google Scholar] [CrossRef]
- Bravarenko, N.I.; Onufriev, M.V.; Stepanichev, M.Y.; Ierusalimsky, V.N.; Balaban, P.M.; Gulyaeva, N.V. Caspase-like activity is essential for long-term synaptic plasticity in the terrestrial snail Helix. Eur. J. Neurosci. 2006, 23, 129–140. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef] [PubMed]
- Madeo, G.; Schirinzi, T.; Martella, G.; Latagliata, E.C.; Puglisi, F.; Shen, J.; Valente, E.M.; Federici, M.; Mercuri, N.B.; Puglisi-Allegra, S.; et al. PINK1 heterozygous mutations induce subtle alterations in dopamine-dependent synaptic plasticity. Mov. Disord. 2014, 29, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Madeo, G.; Martella, G.; Schirinzi, T.; Ponterio, G.; Shen, J.; Bonsi, P.; Pisani, A. Aberrant striatal synaptic plasticity in monogenic parkinsonisms. Neuroscience 2012, 211, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Martella, G.; Maltese, M.; Nisticò, R.; Schirinzi, T.; Madeo, G.; Sciamanna, G.; Ponterio, G.; Tassone, A.; Mandolesi, G.; Vanni, V.; et al. Regional specificity of synaptic plasticity deficits in a knock-in mouse model of DYT1 dystonia. Neurobiol. Dis. 2014, 65, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Martella, G.; Madeo, G.; Maltese, M.; Vanni, V.; Puglisi, F.; Ferraro, E.; Schirinzi, T.; Valente, E.M.; Bonanni, L.; Shen, J.; et al. Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol. Dis. 2016, 91, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Martella, G.; Tassone, A.; Sciamanna, G.; Platania, P.; Cuomo, D.; Viscomi, M.T.; Bonsi, P.; Cacci, E.; Biagioni, S.; Usiello, A.; et al. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: Role of endogenous acetylcholine. Brain 2009, 132, 2336–2349. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar] [CrossRef]
- Kitada, T.; Pisani, A.; Karouani, M.; Haburcak, M.; Martella, G.; Tscherter, A.; Platania, P.; Wu, B.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of parkin-/- mice. J. Neurochem. 2009, 110, 613–621. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sara, Y.; Mozhayeva, M.G.; Liu, X.; Kavalali, E.T. Fast vesicle recycling supports neurotransmission during sustained stimulation at hippocampal synapses. J. Neurosci. 2002, 22, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lovinger, D.M. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc. Natl. Acad. Sci. USA 1997, 94, 2665–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avelar, A.J.; Juliano, S.A.; Garris, P.A. Amphetamine augments vesicular dopamine release in the dorsal and ventral striatum through different mechanisms. J. Neurochem. 2013, 125, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Underhill, S.M.; Wheeler, D.S.; Li, M.; Watts, S.D.; Ingram, S.L.; Amara, S.G. Amphetamine modulates excitatory neurotransmission through endocytosis of the glutamate transporter EAAT3 in dopamine neurons. Neuron 2014, 83, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.S.; Ho, L.B.; Tolliver, B.K.; Wolkowitz, O.M.; Berger, S.P. Partial reversal of stress-induced behavioral sensitization to amphetamine following metyraponetreatment. Brain Res. 1998, 783, 133–142. [Google Scholar] [CrossRef]
- Kalivas, P.W. Cocaine and amphetamine-like psychostimulants: Neurocircuitry and glutamate neuroplasticity. Dialogues Clin. Neurosci. 2007, 9, 389–397. [Google Scholar] [PubMed]
- Li, Z.; Sheng, M. Caspases in synaptic plasticity. Mol. Brain 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Li, Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 2011, 70, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N. BAD: Undertaker by night, candyman by day. Oncogene 2008, 27, S53–S70. [Google Scholar] [CrossRef]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.A.; Valente, E.M. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Hoell, P.; Ahlemeyer, B.; Krieglstein, J. PTEN: A crucial mediator of mitochondria-dependent apoptosis. Apoptosis 2006, 11, 197–207. [Google Scholar] [CrossRef]
- Matsuda, S.; Kitagishi, Y.; Kobayashi, M. Function and characteristics of PINK1 in mitochondria. Oxid. Med. Cell Longev. 2013, 2013, 601587. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Calipari, E.S.; Ferris, M.J. Amphetamine mechanisms and actions at the dopamine terminal revisited. J. Neurosci. 2013, 33, 8923–8925. [Google Scholar] [CrossRef] [PubMed]
- Nash, J.F.; Yamamoto, B.K. Effect of D-amphetamine on the extracellular concentrations of glutamate and dopamine in iprindole-treated rats. Brain Res. 1993, 627, 1–8. [Google Scholar] [CrossRef]
- Del Arco, A.; González-Mora, J.L.; Armas, V.R.; Mora, F. Amphetamine increases the extracellular concentration of glutamate in striatum of the awake rat: Involvement of high affinity transporter mechanisms. Neuropharmacology 1999, 38, 943–954. [Google Scholar] [CrossRef]
- Rawls, S.M.; McGinty, J.F. Delta opioid receptors regulate calcium-dependent, amphetamine-evoked glutamate levels in the rat striatum: An in vivo microdialysis study. Brain Res. 2000, 861, 296–304. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Rego, A.C.; Cardoso, S.M.; Borges, F.; Swerdlow, R.H.; Macedo, T.; de Oliveira, C.R. Mitochondrial dysfunction and caspase activation in rat cortical neurons treated with cocaine or amphetamine. Brain Res. 2006, 1089, 44–54. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Z.; Winterer, J.; Müller, W.; Stanton, P.K. NMDA-dependent, but not group I metabotropic glutamate receptor-dependent, long-term depression at Schaffer collateral-CA1 synapses is associated with long-term reduction of release from the rapidly recycling presynaptic vesicle pool. J. Neurosci. 2006, 26, 10270–10280. [Google Scholar] [CrossRef]
- Sciamanna, G.; Tassone, A.; Mandolesi, G.; Puglisi, F.; Ponterio, G.; Martella, G.; Madeo, G.; Bernardi, G.; Standaert, D.G.; Bonsi, P.; et al. Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J. Neurosci. 2012, 32, 11991–12004. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, P.; Fassio, A.; Valtorta, F.; Benfenati, F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J. Neurosci. 2007, 27, 13520–13531. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.F.; Williams, J.H. Discharge of the readily releasable pool with action potentials at hippocampal synapses. J. Neurophysiol. 2007, 98, 3221–3229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, X.M.; Meinkoth, J.; Pittmanm, R.N. Akt regulates cell survival and apoptosis at a postmitochondrial level. J. Cell Biol. 2000, 151, 483–494. [Google Scholar] [CrossRef]
- Luyet, C.; Schulze, K.; Sayar, B.S.; Howald, D.; Müller, E.J.; Galichet, A. Preclinical studies identify non-apoptotic low-level caspase-3 as therapeutic target in pemphigus vulgaris. PLoS ONE 2015, 10, e0119809. [Google Scholar] [CrossRef]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PINK1+/+ | PINK1+/− | PINK1−/− | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Saline | Z-Dedvd-fmk | PETCM | Saline | Z-Dedvd-fmk | PETCM | Saline | Z-Dedvd-fmk | PETCM | |
| RMP (mV) | −85.2 ± 3.8 | −90.5 ± 1.6 | −89.5 ± 1.3 | −87.2 ± 3.6 | −88.5 ±1.6 | −89.3 ± 1.7 | −79.4 ± 4.0 | −87.10 ± 3.1 | −87.4 ± 2.2 |
| AP amplitude (mV) | 77.0 ± 3.8 | 75.8 ± 1.88 | 76.8 ± 3.03 | 78.4 ± 3.6 | 75.0 ± 2.1 | 73.1 ± 2.9 | 79.4 ± 4.0 | 69.5 ± 4.3 | 75.3 ± 2.5 |
| Membrane resistance (MΩ) | 35.2 ± 2.4 | 36.8 ± 1.7 | 38.2 ± 1.8 | 38.0 ± 2.0 | 38.9 ± 0.7 | 38.7 ± 1.3 | 35.2 ± 3.3 | 34.1 ± 1.2 | 35.6 ± 1.1 |
| Delay to first spike (ms) | 40.5 ± 1.99 | 41.2 ± 1.3 | 42.3 ± 1.6 | 42.1 ± 1.6 | 43.8 ± 1.6 | 42.9 ± 1.5 | 44.2 ± 2.0 | 44.7 ± 1.8 | 46.0 ± 0.3 |
| Rheobase (pA) | 421 ± 12 | 418 ± 9 | 444 ± 11 | 400 ± 18 | 373 ± 6 | 387 ± 12 | 380 ± 18 | 376 ± 6 | 366 ± 15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imbriani, P.; Tassone, A.; Meringolo, M.; Ponterio, G.; Madeo, G.; Pisani, A.; Bonsi, P.; Martella, G. Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 3407. https://doi.org/10.3390/ijms20143407
Imbriani P, Tassone A, Meringolo M, Ponterio G, Madeo G, Pisani A, Bonsi P, Martella G. Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2019; 20(14):3407. https://doi.org/10.3390/ijms20143407
Chicago/Turabian StyleImbriani, Paola, Annalisa Tassone, Maria Meringolo, Giulia Ponterio, Graziella Madeo, Antonio Pisani, Paola Bonsi, and Giuseppina Martella. 2019. "Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease" International Journal of Molecular Sciences 20, no. 14: 3407. https://doi.org/10.3390/ijms20143407
APA StyleImbriani, P., Tassone, A., Meringolo, M., Ponterio, G., Madeo, G., Pisani, A., Bonsi, P., & Martella, G. (2019). Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences, 20(14), 3407. https://doi.org/10.3390/ijms20143407