Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tricellular Tight Junction Proteins and Cancer
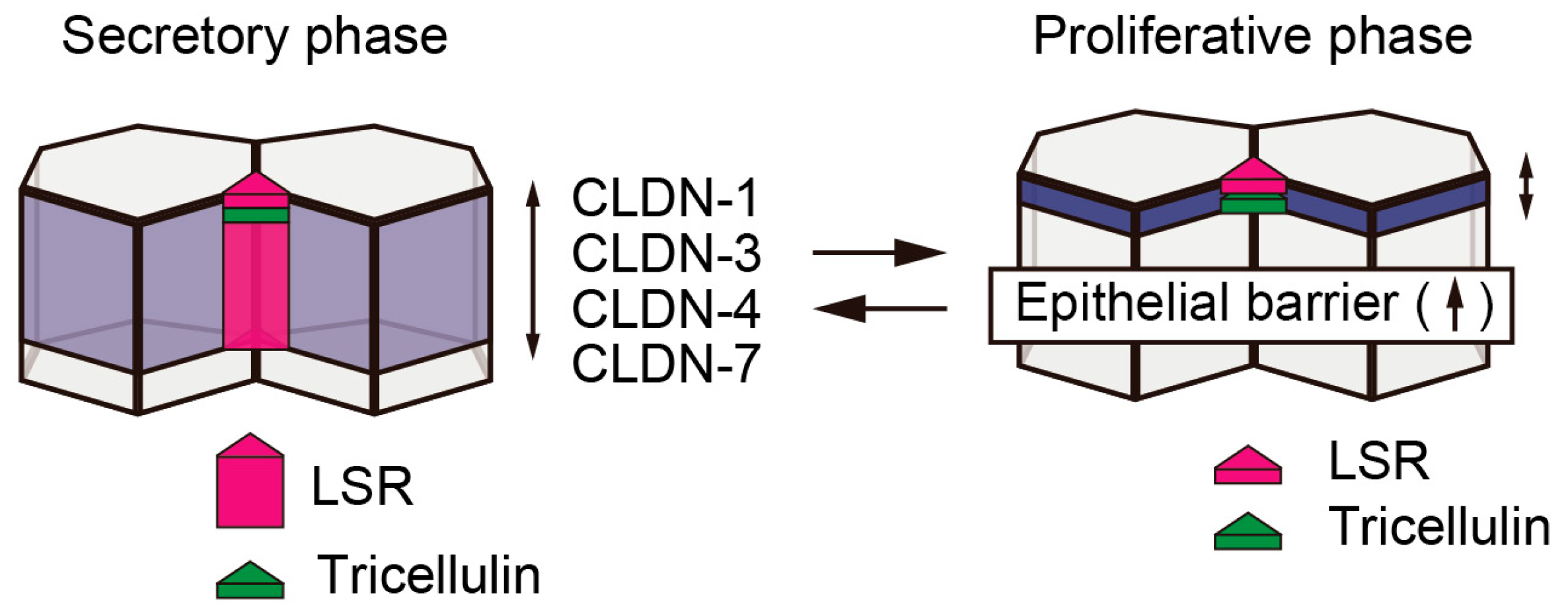
3. Expression and Localization of the Tricellular Tight Junction Proteins, LSR and Tricellulin, during Endometriosis and Endometrial Carcinoma
4. Obesity and Endometrial Cancer
5. Glucose Metabolism and Endometrial Cancer
6. Mechanisms of Enhancement of Cell Invasion Caused by Decreased LSR Expression
7. Hippo Pathway and Endometrial Cancer
8. Crosstalk between the Hippo Pathway and Tight Junctions
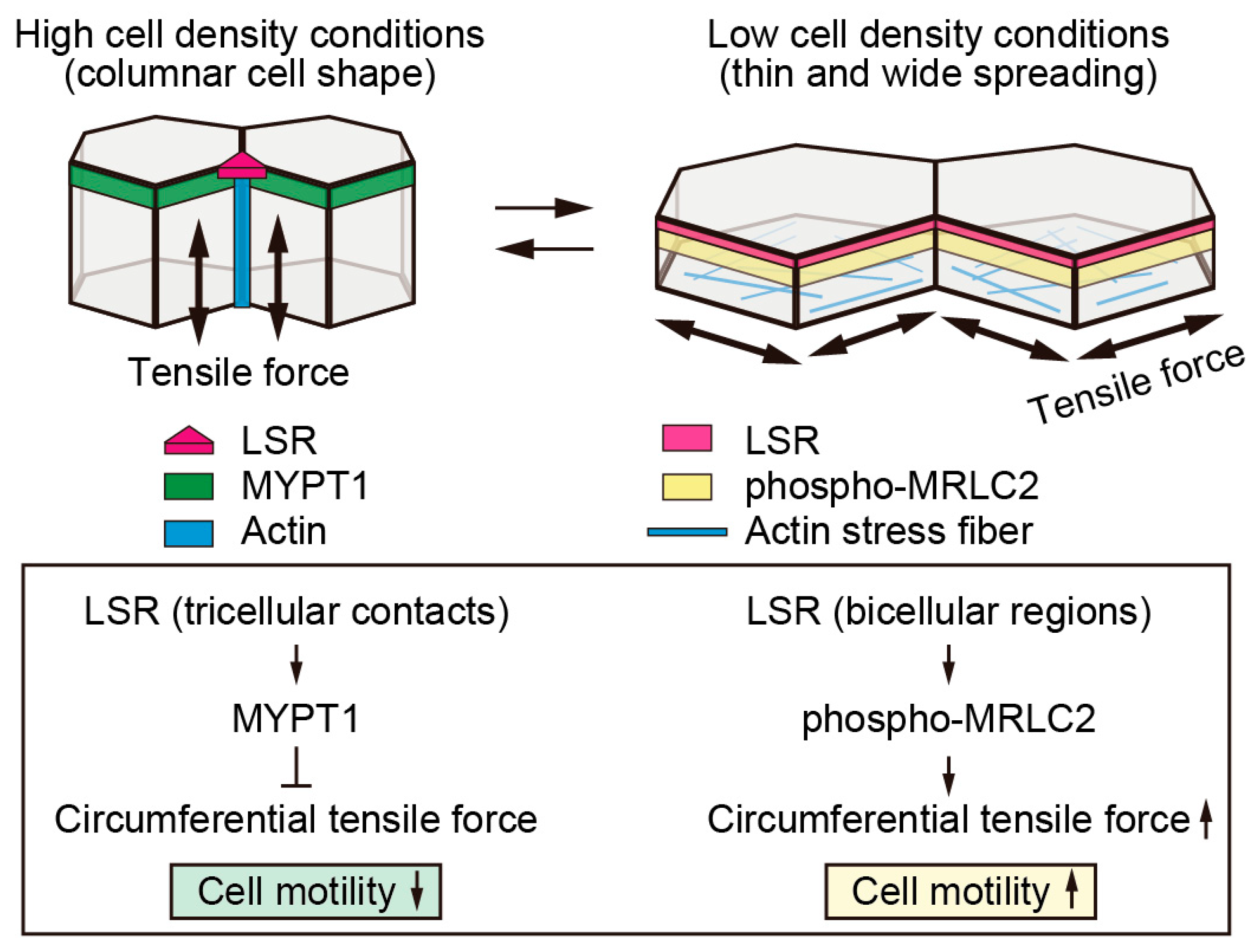
9. Changes in LSR Localization Are Associated with Changes in Cell Size Following Changes in Cell Density
10. Decreased Cellular Tension Causes LSR to Localize at Tricellular Contacts
11. Crosstalk between Intracellular Tension and Cell Junctions
12. Hippo Pathway and Cytoskeletal Dynamics
13. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CLDNs | claudins |
| EMT | epithelial-mesenchymal transition |
| HCC | hepatocellular carcinoma |
| HNSCC | head and neck squamous cell carcinoma |
| iCCC | intrahepatic cholangiocarcinoma |
| LSR | lipolysis-stimulated lipoprotein receptor |
| MLCK | myosin light chain kinase |
| MYPT1 | myosin phosphatase target subunit 1 |
| MRLC2 | myosin regulatory light chain 2 |
| OCLN | occludin |
| OSC | oral squamous cell carcinoma |
| TER | transepithelial electrical resistance |
References
- Garcia, M.A.; Nelson, W.J.; Chavez, N. Cell-cell junctions organize structural and signaling networks. Cold Spring Harb. Perspect. Biol. 2018, 10, a029181. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Izumi, Y.; Oda, Y.; Higashi, T.; Iwamoto, N. Molecular organization of tricellular tight junctions. Tissue Barriers 2014, 2, e28960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukita, S.; Yamazaki, Y.; Katsuno, T.; Tamura, A.; Tsukita, S. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene 2008, 27, 6930–6938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Förster, C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008, 130, 55–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Someya, M.; Kojima, T.; Ogawa, M.; Ninomiya, T.; Nomura, K.; Takasawa, A.; Murata, M.; Tanaka, S.; Saito, T.; Sawada, N. Regulation of tight junctions by sex hormones in normal human endometrial epithelial cells and uterus cancer cell line Sawano. Cell Tissue Res. 2013, 354, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Oda, Y.; Higashi, T.; Iwamoto, N.; Masuda, S. Lipolysis-stimulated lipoprotein receptor: A novel membrane protein of tricellular tight junctions. Ann. N. Y. Acad. Sci. 2012, 1257, 54–58. [Google Scholar] [CrossRef]
- Shimada, H.; Satohisa, S.; Kohno, T.; Takahashi, S.; Hatakeyama, T.; Konno, T.; Tsujiwaki, M.; Saito, T.; Kojima, T. The roles of tricellular tight junction protein lipolysis-stimulated lipoprotein receptor in malignancy of human endometrial cancer cells. Oncotarget 2016, 7, 27735–27752. [Google Scholar] [CrossRef] [Green Version]
- Bilyk, O.; Coatham, M.; Jewer, M.; Postovit, L.M. Epithelial-to-Mesenchymal Transition in the female reproductive tract: From normal functioning to disease pathology. Front. Oncol. 2017, 7, 1–21. [Google Scholar] [CrossRef]
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4. [Google Scholar] [CrossRef]
- Martin, T.A.; Jiang, W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta Biomembr. 2009, 1788, 872–891. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.A. The role of tight junctions in cancer metastasis. Semin. Cell Dev. Biol. 2014, 36, 224–231. [Google Scholar] [CrossRef]
- Leech, A.O.; Cruz, R.G.B.; Hill, A.D.K.; Hopkins, A.M. Paradigms lost—An emerging role for over-expression of tight junction adhesion proteins in cancer pathogenesis. Ann. Transl. Med. 2015, 3, 1–15. [Google Scholar]
- Kondoh, A.; Takano, K.I.; Kojima, T.; Ohkuni, T.; Kamekura, R.; Ogasawara, N.; Go, M.; Sawada, N.; Himi, T. Altered expression of claudin-1, claudin-7, and tricellulin regardless of human papilloma virus infection in human tonsillar squamous cell carcinoma. Acta Otolaryngol. 2011, 131, 861–868. [Google Scholar] [CrossRef]
- Patonai, A.; Erdélyi-Belle, B.; Korompay, A.; Somorácz, A.; Straub, B.K.; Schirmacher, P.; Kovalszky, I.; Lotz, G.; Kiss, A.; Schaff, Z. Claudins and tricellulin in fibrolamellar hepatocellular carcinoma. Virchows Arch. 2011, 458, 679–688. [Google Scholar] [CrossRef]
- Somorácz, A.; Korompay, A.; Törzsök, P.; Patonai, A.; Erdélyi-Belle, B.; Lotz, G.; Schaff, Z.; Kiss, A. Tricellulin expression and its prognostic significance in primary liver carcinomas. Pathol. Oncol. Res. 2014, 20, 755–764. [Google Scholar] [CrossRef]
- Korompay, A.; Borka, K.; Lotz, G.; Somorácz, A.; Törzsök, P.; Erdélyi-Belle, B.; Kenessey, I.; Baranyai, Z.; Zsoldos, F.; Kupcsulik, P.; et al. Tricellulin expression in normal and neoplastic human pancreas. Histopathology 2012, 60, E76–E86. [Google Scholar] [CrossRef]
- Masuda, R.; Semba, S.; Mizuuchi, E.; Yanagihara, K.; Yokozaki, H. Negative regulation of the tight junction protein tricellulin by snail-induced epithelial-mesenchymal transition in gastric carcinoma cells. Pathobiology 2010, 77, 106–113. [Google Scholar] [CrossRef]
- Reaves, D.K.; Fagan-Solis, K.D.; Dunphy, K.; Oliver, S.D.; Scott, D.W.; Fleming, J.M. The role of lipolysis stimulated lipoprotein receptor in breast cancer and directing breast cancer cell behavior. PLoS ONE 2014, 9, e91747. [Google Scholar] [CrossRef]
- García, J.M.; Peña, C.; García, V.; Domínguez, G.; Muñoz, C.; Silva, J.; Millán, I.; Diaz, R.; Lorenzo, Y.; Rodriguez, R.; et al. Prognostic value of LISCH7 mRNA in plasma and tumor of colon cancer patients. Clin. Cancer Res. 2007, 13, 6351–6358. [Google Scholar] [CrossRef]
- Takano, K.; Kakuki, T.; Obata, K.; Nomura, K.; Miyata, R.; Kondo, A.; Kurose, M.; Kakiuchi, A.; Kaneko, Y.; Kohno, T.; et al. The behavior and role of lipolysis-stimulated lipoprotein receptor, a component of tricellular tight junctions, in head and neck squamous cell carcinomas. Anticancer Res. 2016, 36, 5895–5904. [Google Scholar] [CrossRef]
- Sappayatosok, K.; Phattarataratip, E. Overexpression of claudin-1 is associated with advanced clinical stage and invasive pathologic characteristics of oral squamous cell carcinoma. Head Neck Pathol. 2015, 9, 173–180. [Google Scholar] [CrossRef]
- Li, W.; Dong, Q.; Li, L.; Zhang, Z.; Cai, X.; Pan, X. Prognostic significance of claudin-1 and cyclin B1 protein expression in patients with hypopharyngeal squamous cell carcinoma. Oncol. Lett. 2016, 11, 2995–3002. [Google Scholar] [CrossRef] [Green Version]
- Grund, S.; Grümmer, R. Direct cell-cell interactions in the endometrium and in endometrial pathophysiology. Int. J. Mol. Sci. 2018, 19, 2227. [Google Scholar] [CrossRef]
- Pan, X.Y.; Wang, B.; Che, Y.C.; Weng, Z.P.; Dai, H.Y.; Peng, W. Expression of claudin-3 and claudin-4 in normal, hyperplastic, and malignant endometrial tissue. Int. J. Gynecol. Cancer 2007, 17, 233–241. [Google Scholar] [CrossRef]
- Konecny, G.E.; Agarwal, R.; Keeney, G.A.; Winterhoff, B.; Jones, M.B.; Mariani, A.; Riehle, D.; Neuper, C.; Dowdy, S.C.; Wang, H.J.; et al. Claudin-3 and claudin-4 expression in serous papillary, clear-cell, and endometrioid endometrial cancer. Gynecol. Oncol. 2008, 109, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Lortet-Tieulent, J.; Ferlay, J.; Bray, F.; Jemal, A. International patterns and trends in endometrial cancer incidence, 1978–2013. J. Natl. Cancer Inst. 2018, 110, 354–361. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and cancer mechanisms: Cancer metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef]
- Byrne, F.L.; Poon, I.K.H.; Modesitt, S.C.; Tomsig, J.L.; Chow, J.D.Y.; Healy, M.E.; Baker, W.D.; Atkins, K.A.; Lancaster, J.M.; Marchion, D.C.; et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014, 74, 5832–5845. [Google Scholar] [CrossRef]
- Gelsomino, L.; Naimo, G.D.; Catalano, S.; Mauro, L.; Andò, S. The emerging role of adiponectin in female malignancies. Int. J. Mol. Sci. 2019, 20, 2127. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Ashizawa, N.; Yahata, T.; Quan, J.; Adachi, S.; Yoshihara, K.; Tanaka, K. Serum leptin-adiponectin ratio and endometrial cancer risk in postmenopausal female subjects. Gynecol. Oncol. 2010, 119, 65–69. [Google Scholar] [CrossRef]
- Gong, T.T.; Wu, Q.J.; Wang, Y.L.; Ma, X.X. Circulating adiponectin, leptin and adiponectin-leptin ratio and endometrial cancer risk: Evidence from a meta-analysis of epidemiologic studies. Int. J. Cancer 2015, 137, 1967–1978. [Google Scholar] [CrossRef]
- Dupont, J.Ë.; Reverchon, M.; Cloix, L.; Froment, P.; Ramé, C. Involvement of adipokines, AMPK, PI3K and the PPAR signaling pathways in ovarian follicle development and cancer. Int. J. Dev. Biol. 2012, 56, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin action in normal and pathological pregnancies. J. Cell. Mol. Med. 2018, 22, 716–727. [Google Scholar] [CrossRef]
- Fan, Y.; Gan, Y.; Shen, Y.; Cai, X.; Song, Y.; Zhao, F.; Yao, M.; Gu, J.; Tu, H. Leptin signaling enhances cell invasion and promotes the metastasis of human pancreatic cancer via increasing MMP-13 production. Oncotarget 2015, 6, 16120–16134. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Choi, Y.S.; Choi, J.H. Leptin promotes human endometriotic cell migration and invasion by up-regulating MMP-2 through the JAK2/STAT3 signaling pathway. Mol. Hum. Reprod. 2015, 21, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Handy, J.A.; Fu, P.P.; Kumar, P.; Mells, J.E.; Sharma, S.; Saxena, N.K.; Anania, F.A. Adiponectin inhibits leptin signalling via multiple mechanisms to exert protective effects against hepatic fibrosis. Biochem. J. 2011, 440, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Stenger, C.; Hanse, M.; Pratte, D.; Mbala, M.L.; Akbar, S.; Koziel, V.; Escanyé, M.C.; Kriem, B.; Malaplate-Armand, C.; Olivier, J.L.; et al. Up-regulation of hepatic lipolysis stimulated lipoprotein receptor by leptin: A potential lever for controlling lipid clearance during the postprandial phase. FASEB J. 2010, 24, 4218–4228. [Google Scholar] [CrossRef]
- Yen, F.T.; Roitel, O.; Bonnard, L.; Notet, V.; Pratte, D.; Stenger, C.; Magueur, E.; Bihain, B.E. Lipolysis stimulated lipoprotein receptor: A novel molecular link between hyperlipidemia, weight gain, and atherosclerosis in mice. J. Biol. Chem. 2008, 283, 25650–25659. [Google Scholar] [CrossRef]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, obesity, and cancer: Clash of the bigwigs in health and disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef]
- Lee, T.Y.; Martinez-Outschoorn, U.E.; Schilder, R.J.; Kim, C.H.; Richard, S.D.; Rosenblum, N.G.; Johnson, J.M. Metformin as a therapeutic target in endometrial cancers. Front. Oncol. 2018, 8, 341. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, C.; Ying, Y.; Luo, L.; Huang, D.; Luo, Z. Metformin and berberine, two versatile drugs in treatment of common metabolic diseases. Oncotarget 2018, 9, 10135–10146. [Google Scholar] [CrossRef]
- Umene, K.; Banno, K.; Kisu, I.; Yanokura, M.; Nogami, Y.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; et al. New candidate therapeutic agents for endometrial cancer: Potential for clinical practice (Review). Oncol. Rep. 2013, 29, 855–860. [Google Scholar] [CrossRef]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Canfield, P.E.; Geerdes, A.M.; Molitoris, B.A. Effect of reversible ATP depletion on tight-junction integrity in LLC-PK1 cells. Am. J. Physiol. Physiol. 2017, 261, F1038–F1045. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. USA 2006, 103, 17272–17277. [Google Scholar] [CrossRef] [Green Version]
- Aznar, N.; Patel, A.; Rohena, C.C.; Dunkel, Y.; Joosen, L.P.; Taupin, V.; Kufareva, I.; Farquhar, M.G.; Ghosh, P. AMP-activated protein kinase fortifies epithelial tight junctions during energetic stress via its effector GIV/Girdin. eLife 2016, 5, 1–33. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Zou, J.; Hong, L.; Luo, C.; Li, Z.; Zhu, Y.; Huang, T.; Zhang, Y.; Yuan, H.; Hu, Y.; Wen, T.; et al. Metformin inhibits estrogen-dependent endometrial cancer cell growth by activating the AMPK–FOXO1 signal pathway. Cancer Sci. 2016, 107, 1806–1817. [Google Scholar] [CrossRef]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Dufresne, J.; Cyr, D.G. Activation of an SP binding site is crucial for the expression of claudin 1 in rat epididymal principal cells. Biol. Reprod. 2007, 76, 825–832. [Google Scholar] [CrossRef]
- Honda, H.; Pazin, M.J.; Ji, H.; Wernyj, R.P.; Morin, P.J. Crucial roles of Sp1 and epigenetic modifications in the regulation of the cldn4 promoter in ovarian cancer cells. J. Biol. Chem. 2006, 281, 21433–21444. [Google Scholar] [CrossRef]
- Luk, J.M.; Tong, M.K.; Mok, B.W.; Tam, P.C.; Yeung, W.S.B.; Lee, K.F. Sp1 site is crucial for the mouse claudin-19 gene expression in the kidney cells. FEBS Lett. 2004, 578, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Shimada, H.; Satohisa, S.; Kohno, T.; Konno, T.; Arimoto, C.; Saito, T.; Kojima, T. Downregulation of lipolysis-stimulated lipoprotein receptor promotes cell invasion via claudin-1-mediated matrix metalloproteinases in human endometrial cancer. Oncol. Lett. 2017, 16, 6776–6782. [Google Scholar] [CrossRef]
- Oku, N.; Sasabe, E.; Ueta, E.; Yamamoto, T.; Osaki, T. Tight junction protein claudin-1 enhances the invasive activity of oral squamous cell carcinoma cells by promoting cleavage of laminin-5 γ2 chain via matrix metalloproteinase (MMP)-2 and membrane-type MMP-1. Cancer Res. 2006, 66, 5251–5257. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Fanning, A.S.; Anderson, J.M.; Boireau, S.; Samuel, M.S.; Pannequin, J.; Ryan, J.L.; Choquet, A.; Chapuis, H.; Rebillard, X.; et al. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [Green Version]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef]
- Brown, G.T.; Murray, G.I. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2015, 237, 273–281. [Google Scholar] [CrossRef]
- Itoh, Y.; Seiki, M. MT1-MMP: A potent modifier of pericellular microenvironment. J. Cell. Physiol. 2006, 206, 1–8. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Bao, Y.; Nakagawa, K.; Yang, Z.; Ikeda, M.; Withanage, K.; Ishigami-Yuasa, M.; Okuno, Y.; Hata, S.; Nishina, H.; Hata, Y. A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J. Biochem. 2011, 150, 199–208. [Google Scholar] [CrossRef]
- Zheng, H.X.; Wu, L.N.; Xiao, H.; Du, Q.; Liang, J.F. Inhibitory effects of dobutamine on human gastric adenocarcinoma. World J. Gastroenterol. 2014, 20, 17092–17099. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase-an energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Shimada, H.; Abe, S.; Kohno, T.; Satohisa, S.; Konno, T.; Takahashi, S.; Hatakeyama, T.; Arimoto, C.; Kakuki, T.; Kaneko, Y.; et al. Loss of tricellular tight junction protein LSR promotes cell invasion and migration via upregulation of TEAD1/AREG in human endometrial cancer. Sci. Rep. 2017, 7, 37049. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.; Giancotti, F.G. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014, 588, 2743–2752. [Google Scholar] [CrossRef]
- Gladden, A.B.; Hebert, A.M.; Schneeberger, E.E.; McClatchey, A.I. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev. Cell 2010, 19, 727–739. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Zhou, Y.; Zhang, J.; Cheng, A.S.L.; Yu, J.; To, K.F.; Kang, W. The physiological role of Motin family and its dysregulation in tumorigenesis. J. Transl. Med. 2018, 16, 98. [Google Scholar] [CrossRef]
- Ortiz, A.; Lee, Y.C.; Yu, G.; Liu, H.C.; Lin, S.C.; Bilen, M.A.; Cho, H.; Yu-Lee, L.Y.; Lin, S.H. Angiomotin is a novel component of cadherin-11/β-catenin/p120 complex and is critical for cadherin-11-mediated cell migration. FASEB J. 2015, 29, 1080–1091. [Google Scholar] [CrossRef]
- Hakami, F.; Darda, L.; Stafford, P.; Woll, P.; Lambert, D.W.; Hunter, K.D. The roles of HOXD10 in the development and progression of head and neck squamous cell carcinoma (HNSCC). Br. J. Cancer 2014, 111, 807–816. [Google Scholar] [CrossRef] [Green Version]
- Levchenko, T.; Bratt, A.; Arbiser, J.L.; Holmgren, L. Angiomotin expression promotes hemangioendothelioma invasion. Oncogene 2004, 23, 1469–1473. [Google Scholar] [CrossRef] [Green Version]
- Couderc, C.; Boin, A.; Fuhrmann, L.; Vincent-Salomon, A.; Mandati, V.; Kieffer, Y.; Mechta-Grigoriou, F.; Del Maestro, L.; Chavrier, P.; Vallerand, D.; et al. AMOTL1 promotes breast cancer progression and is antagonized by Merlin. Neoplasia 2016, 18, 10–24. [Google Scholar] [CrossRef]
- Wan, H.Y.; Li, Q.Q.; Zhang, Y.; Tian, W.; Li, Y.N.; Liu, M.; Li, X.; Tang, H. MiR-124 represses vasculogenic mimicry and cell motility by targeting AmotL1 in cervical cancer cells. Cancer Lett. 2014, 355, 148–158. [Google Scholar] [CrossRef]
- Mojallal, M.; Zheng, Y.; Hultin, S.; Audebert, S.; van Harn, T.; Johnsson, P.; Lenander, C.; Fritz, N.; Mieth, C.; Corcoran, M.; et al. AmotL2 disrupts apical–basal cell polarity and promotes tumour invasion. Nat. Commun. 2014, 5, 4557. [Google Scholar] [CrossRef]
- Artinian, N.; Cloninger, C.; Holmes, B.; Benavides-Serrato, A.; Bashir, T.; Gera, J. Phosphorylation of the Hippo pathway component AMOTL2 by the mTORC2 kinase promotes YAP signaling, resulting in enhanced glioblastoma growth and invasiveness. J. Biol. Chem. 2015, 290, 19387–19401. [Google Scholar] [CrossRef]
- Li, W.; Cooper, J.; Karajannis, M.A.; Giancotti, F.G. Merlin: A tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 2012, 13, 204–215. [Google Scholar] [CrossRef]
- Yi, C.; Troutman, S.; Fera, D.; Stemmer-Rachamimov, A.; Avila, J.L.; Christian, N.; Persson, N.L.; Shimono, A.; Speicher, D.W.; Marmorstein, R.; et al. A tight junction-associated Merlin-Angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 2011, 19, 527–540. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Demidenko, Z.N.; Blagosklonny, M. V Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc. Natl. Acad. Sci. USA 2014, 111, 8832–8837. [Google Scholar] [CrossRef]
- Gumbiner, B.M.; Kim, N.-G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Kikuchi, S.; Ninomiya, T.; Kojima, T. The bicellular tensile force sorts the localization of LSRs in bicellular and tricellular junctions. Ann. N. Y. Acad. Sci. 2017, 1397, 185–194. [Google Scholar] [CrossRef]
- Levayer, R.; Lecuit, T. Biomechanical regulation of contractility: Spatial control and dynamics. Trends Cell Biol. 2012, 22, 61–81. [Google Scholar] [CrossRef]
- Gutzman, J.H.; Sive, H. Epithelial relaxation mediated by the myosin phosphatase regulator Mypt1 is required for brain ventricle lumen expansion and hindbrain morphogenesis. Development 2010, 137, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Mui, K.L.; Chen, C.S.; Assoian, R.K. The mechanical regulation of integrin-cadherin crosstalk organizes cells, signaling and forces. J. Cell Sci. 2016, 129, 1093–1100. [Google Scholar] [CrossRef]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Semba, S.; Khan, R.I.; Bochimoto, H.; Watanabe, T.; Fujiya, M.; Kohgo, Y.; Liu, Y.; Taniguchi, T. Focal adhesion kinase regulates intestinal epithelial barrier function via redistribution of tight junction. Biochim. Biophys. Acta 2013, 1832, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Siu, E.R.; Wong, E.W.P.; Mruk, D.D.; Porto, C.S.; Cheng, C.Y. Focal adhesion kinase is a blood-testis barrier regulator. Proc. Natl. Acad. Sci. USA 2009, 106, 9298–9303. [Google Scholar] [CrossRef]
- Oda, Y.; Otani, T.; Ikenouchi, J.; Furuse, M. Tricellulin regulates junctional tension of epithelial cells at tricellular contacts via Cdc42. J. Cell Sci. 2014, 127, 4201–4212. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. ROCKs: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef]
- Kale, V.P.; Hengst, J.A.; Desai, D.H.; Dick, T.E.; Choe, K.N.; Colledge, A.L.; Takahashi, Y.; Sung, S.; Amin, S.G.; Yun, J.K. A novel selective multikinase inhibitor of ROCK and MRCK effectively blocks cancer cell migration and invasion. Cancer Lett. 2014, 354, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Nakano, T.; Erdodi, F.; Hartshorne, D.J. Myosin phosphatase: Structure, regulation and function. Mol. Cell. Biochem. 2004, 259, 197–209. [Google Scholar] [CrossRef]
- Masuda, S.; Oda, Y.; Sasaki, H.; Ikenouchi, J.; Higashi, T.; Akashi, M.; Nishi, E.; Furuse, M. LSR defines cell corners for tricellular tight junction formation in epithelial cells. J. Cell Sci. 2011, 124, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, D.; Kano, F.; Taguchi, Y.; Sugawara, T.; Nishizono, T.; Nishikawa, K.; Oda, Y.; Furuse, M.; Murata, M. JNK1/2-dependent phosphorylation of angulin-1/LSR is required for the exclusive localization of angulin-1/LSR and tricellulin at tricellular contacts in EpH4 epithelial sheet. Genes Cells 2014, 19, 565–581. [Google Scholar] [CrossRef]
- Takai, Y.; Nakanishi, H. Nectin and afadin: Novel organizers of intercellular junctions. J. Cell Sci. 2003, 116, 17–27. [Google Scholar] [CrossRef]
- Zhang, J.; Betson, M.; Erasmus, J.; Zeikos, K.; Bailly, M.; Cramer, L.P.; Braga, V.M.M. Actin at cell-cell junctions is composed of two dynamic and functional populations. J. Cell Sci. 2005, 118, 5549–5562. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.W.; Gerstenzang, E.; Ossipova, O.; Sokol, S.Y. Lulu regulates shroom-induced apical constriction during neural tube closure. PLoS ONE 2013, 8, e81854. [Google Scholar] [CrossRef]
- Nishimura, T.; Takeichi, M. Shroom3-mediated recruitment of Rho kinases to the apical cell junctions regulates epithelial and neuroepithelial planar remodeling. Development 2008, 135, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Boulan, E.; Macara, I.G. Organization and execution of the epithelial polarity programme. Nat. Rev. Mol. Cell Biol. 2014, 15, 225–242. [Google Scholar] [CrossRef]
- Toya, M.; Kobayashi, S.; Kawasaki, M.; Shioi, G.; Kaneko, M.; Ishiuchi, T.; Misaki, K.; Meng, W.; Takeichi, M. CAMSAP3 orients the apical-to-basal polarity of microtubule arrays in epithelial cells. Proc. Natl. Acad. Sci. USA 2015, 113, 1–6. [Google Scholar] [CrossRef]
- Bazellières, E.; Massey-Harroche, D.; Barthélémy-Requin, M.; Richard, F.; Arsanto, J.P.; Le Bivic, A. Apico-basal elongation requires a drebrin-E-EB3 complex in columnar human epithelial cells. J. Cell Sci. 2012, 125, 919–931. [Google Scholar] [CrossRef]
- Butkevich, E.; Hülsmann, S.; Wenzel, D.; Shirao, T.; Duden, R.; Majoul, I. Drebrin is a novel connexin-43 binding partner that links gap junctions to the submembrane cytoskeleton. Curr. Biol. 2004, 14, 650–658. [Google Scholar] [CrossRef]
- Guillemot, L.; Paschoud, S.; Pulimeno, P.; Foglia, A.; Citi, S. The cytoplasmic plaque of tight junctions: A scaffolding and signalling center. Biochim. Biophys. Acta Biomembr. 2008, 1778, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Miyake, Y.; Inoue, N.; Nishimura, K.; Kinoshita, N.; Hosoya, H.; Yonemura, S. Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp. Cell Res. 2006, 312, 1637–1650. [Google Scholar] [CrossRef]
- Iden, S.; Collard, J.G. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell Biol. 2008, 9, 846–859. [Google Scholar] [CrossRef]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Porazinski, S.; Wang, H.; Asaoka, Y.; Behrndt, M.; Miyamoto, T.; Morita, H.; Hata, S.; Sasaki, T.; Krens, S.F.G.; Osada, Y.; et al. YAP is essential for tissue tension to ensure vertebrate 3D body shape. Nature 2015, 521, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Tsujiura, M.; Mazack, V.; Sudol, M.; Kaspar, H.G.; Nash, J.; Carey, D.J.; Gogoi, R. Yes-Associated Protein (YAP) modulates oncogenic features and radiation sensitivity in endometrial cancer. PLoS ONE 2014, 9, e100974. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohno, T.; Konno, T.; Kojima, T. Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3555. https://doi.org/10.3390/ijms20143555
Kohno T, Konno T, Kojima T. Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells. International Journal of Molecular Sciences. 2019; 20(14):3555. https://doi.org/10.3390/ijms20143555
Chicago/Turabian StyleKohno, Takayuki, Takumi Konno, and Takashi Kojima. 2019. "Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells" International Journal of Molecular Sciences 20, no. 14: 3555. https://doi.org/10.3390/ijms20143555
APA StyleKohno, T., Konno, T., & Kojima, T. (2019). Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells. International Journal of Molecular Sciences, 20(14), 3555. https://doi.org/10.3390/ijms20143555