Roles of Myosin-Mediated Membrane Trafficking in TGF-β Signaling


 and
and 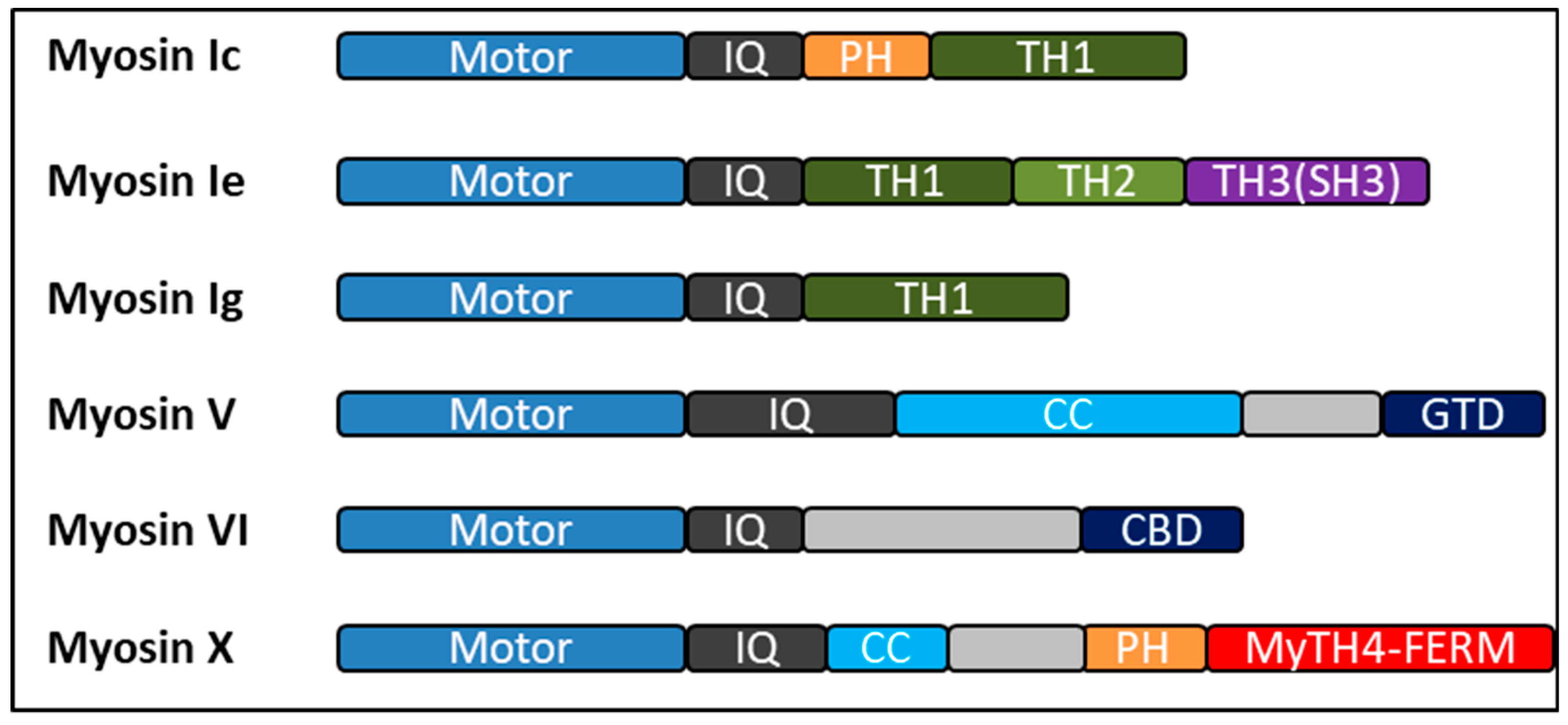{kind=link}
{kind=link}
Abstract
:1. Introduction
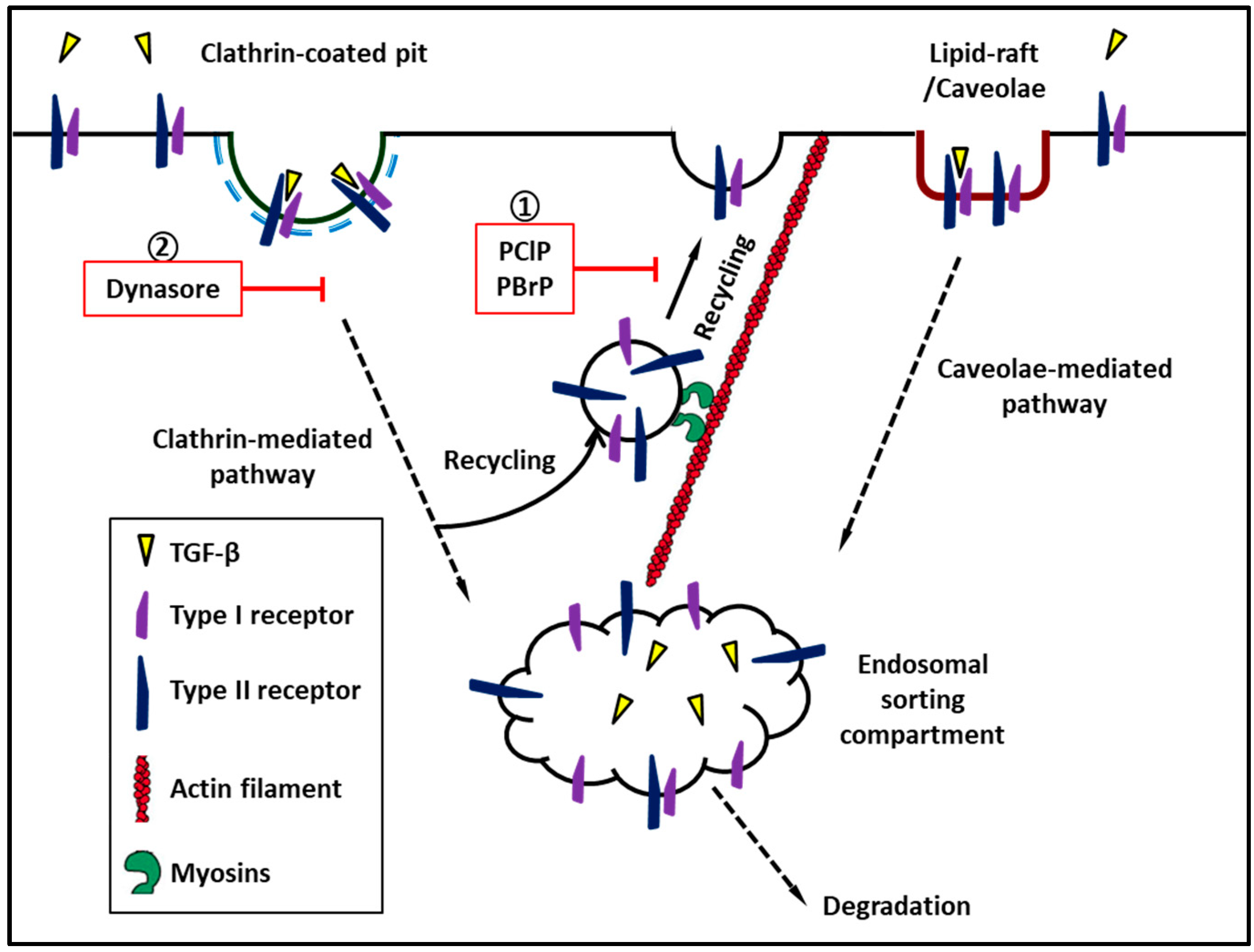
2. Distribution of TGF-β Receptors in the Plasma Membrane
3. TGF-β Signaling Is Modulated by Receptor Trafficking
4. Myosin
5. Myosin I
5.1. Myo1c
5.2. Myo1e
5.3. Myo1g
6. Myosin V
7. Myosin VI
8. Myosin X
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| TGF-β | transforming growth factor-β |
| ECM | extracellular matrix |
| Smad | homologies to the Caenorhabditis elegans SMA (“small” worm phenotype) and Drosophila MAD (“Mothers Against Decapentaplegic”) |
| EMT | epithelial to mesenchymal transition |
| NEDD | neural precursor cell-expressed, developmentally downregulated |
| CCPs | clathrin-coated pits |
| IL-6 | interleukin 6 |
| c-Cbl | casitas B-lineage Lymphoma |
| ADAM12 | a disintegrin and metalloproteinase domain-containing protein 12 |
| CME | clathrin-mediated endocytosis |
| IQ | IQ calmodulin-binding motif |
| PClP | pentachloropseudilin |
| PBrP | pentabromopseudilin |
| PH | pleckstrin homology |
| SH3 | src-homology 3 domain |
| HA | hyaluronic acid |
| ZO-1 | zonula occludens-1 |
| MAPK | mitogen-activated protein kinase |
References
- Kingsley, D.M. The tgf-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, D.B. Latent transforming growth factor-beta (tgf-beta) binding proteins: Orchestrators of tgf-beta availability. J. Biol. Chem. 2005, 280, 7409–7412. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Wotton, D. Transcriptional control by the tgf-β/smad signaling system. Embo. J. 2000, 19, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of tgf-β signaling from cell membrane to the nucleus. cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in tgf-β family signalling. Nature 2003, 425, 577. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Derynck, R. Tgf-β signaling in cancer–a double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [PubMed]
- de Caestecker, M.P.; Piek, E.; Roberts, A.B. Role of transforming growth factor-β signaling in cancer. J. Natl. Cancer Inst. 2000, 92, 1388–1402. [Google Scholar] [CrossRef]
- Hannon, G.J.; Beach, D. Pl5ink4b is a potentia| effector of tgf-β-induced cell cycle arrest. Nature 1994, 371, 257. [Google Scholar] [CrossRef]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.-F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of tgf-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964. [Google Scholar] [CrossRef]
- Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Jakowlew, S.; Alvarez-Mon, M.; Derynck, R.; Sporn, M.B.; Fauci, A.S. Production of transforming growth factor beta by human t lymphocytes and its potential role in the regulation of t cell growth. J. Exp. Med. 1986, 163, 1037–1050. [Google Scholar] [CrossRef]
- Mempel, T.R.; Pittet, M.J.; Khazaie, K.; Weninger, W.; Weissleder, R.; von Boehmer, H.; von Andrian, U.H. Regulatory t cells reversibly suppress cytotoxic t cell function independent of effector differentiation. Immunity 2006, 25, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Bellone, G.; Aste-Amezaga, M.; Trinchieri, G.; Rodeck, U. Regulation of nk cell functions by tgf-beta 1. J. Immunol. 1995, 155, 1066–1073. [Google Scholar]
- Geissmann, F.; Revy, P.; Regnault, A.; Lepelletier, Y.; Dy, M.; Brousse, N.; Amigorena, S.; Hermine, O.; Durandy, A. Tgf-β1 prevents the noncognate maturation of human dendritic langerhans cells. J. Immunol. 1999, 162, 4567–4575. [Google Scholar]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-β controls development, homeostasis, and tolerance of t cells by regulatory t cell-dependent and-independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Piek, E.; Moustakas, A.; Kurisaki, A.; Heldin, C.-H.; ten Dijke, P. Tgf-(beta) type i receptor/alk-5 and smad proteins mediate epithelial to mesenchymal transdifferentiation in nmumg breast epithelial cells. J. Cell Sci. 1999, 112, 4557–4568. [Google Scholar]
- Portella, G.; Cumming, S.A.; Liddell, J.; Cui, W.; Ireland, H.; Akhurst, R.J.; Balmain, A. Transforming growth factor beta is essential for spindle cell conversion of mouse skin carcinoma in vivo: Implications for tumor invasion. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1998, 9, 393–404. [Google Scholar]
- Kitagawa, K.; Murata, A.; Matsuura, N.; Tohya, K.; Takaichi, S.; Monden, M.; Inoue, M. Epithelial-mesenchymal transformation of a newly established cell line from ovarian adenosarcoma by transforming growth factor-b1. Int. J. Cancer 1996, 66, 91–97. [Google Scholar] [CrossRef]
- Janji, B.; Melchior, C.; Gouon, V.; Vallar, L.; Kieffer, N. Autocrine tgf-β-regulated expression of adhesion receptors and integrin-linked kinase in ht-144 melanoma cells correlates with their metastatic phenotype. Int. J. Cancer 1999, 83, 255–262. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. Tgf-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117. [Google Scholar] [CrossRef]
- Derynck, R.; Goeddel, D.V.; Ullrich, A.; Gutterman, J.U.; Williams, R.D.; Bringman, T.S.; Berger, W.H. Synthesis of messenger rnas for transforming growth factors α and β and the epidermal growth factor receptor by human tumors. Cancer Res. 1987, 47, 707–712. [Google Scholar]
- Oft, M.; Heider, K.-H.; Beug, H. Tgfβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 1998, 8, 1243–1252. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. Tgf-β-induced emt: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef]
- Dooley, S.; Ten Dijke, P. Tgf-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Quintanilla, M.; Bernabeu, C. Tgf-beta/tgf-beta receptor system and its role in physiological and pathological conditions. Clin. Sci. (Lond) 2011, 121, 233–251. [Google Scholar] [CrossRef]
- Yan, X.; Chen, Y.G. Posttranslational modifications of tgf-beta receptors. Methods Mol. Biol. 2016, 1344, 49–61. [Google Scholar]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming growth factor-beta receptors and smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- Xu, P.; Liu, J.; Derynck, R. Post-translational regulation of tgf-beta receptor and smad signaling. Febs. Lett 2012, 586, 1871–1884. [Google Scholar] [CrossRef]
- Yakymovych, I.; Yakymovych, M.; Heldin, C.H. Intracellular trafficking of transforming growth factor beta receptors. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 3–11. [Google Scholar] [CrossRef]
- Zhang, X.L.; Topley, N.; Ito, T.; Phillips, A. Interleukin-6 regulation of transforming growth factor (tgf)-beta receptor compartmentalization and turnover enhances tgf-beta1 signaling. J. Biol. Chem. 2005, 280, 12239–12245. [Google Scholar] [CrossRef]
- Ito, T.; Williams, J.D.; Fraser, D.J.; Phillips, A.O. Hyaluronan regulates transforming growth factor-beta1 receptor compartmentalization. J. Biol. Chem. 2004, 279, 25326–25332. [Google Scholar] [CrossRef]
- Atfi, A.; Dumont, E.; Colland, F.; Bonnier, D.; L’Helgoualc’h, A.; Prunier, C.; Ferrand, N.; Clement, B.; Wewer, U.M.; Theret, N. The disintegrin and metalloproteinase adam12 contributes to tgf-beta signaling through interaction with the type ii receptor. J. Cell Biol. 2007, 178, 201–208. [Google Scholar] [CrossRef]
- Zuo, W.; Huang, F.; Chiang, Y.J.; Li, M.; Du, J.; Ding, Y.; Zhang, T.; Lee, H.W.; Jeong, L.S.; Chen, Y.; et al. C-cbl-mediated neddylation antagonizes ubiquitination and degradation of the tgf-beta type ii receptor. Mol. Cell 2013, 49, 499–510. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Razani, B.; Zhang, X.L.; Bitzer, M.; von Gersdorff, G.; Bottinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor (tgf)-beta/smad signaling through an interaction with the tgf-beta type i receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef]
- Luga, V.; McLean, S.; Le Roy, C.; O’Connor-McCourt, M.; Wrana, J.L.; Di Guglielmo, G.M. The extracellular domain of the tgfbeta type ii receptor regulates membrane raft partitioning. Biochem. J. 2009, 421, 119–131. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, Q.; Du, J.; Luo, S.; Xia, J.; Chen, Y.G. Pick1 promotes caveolin-dependent degradation of tgf-beta type i receptor. Cell Res. 2012, 22, 1467–1478. [Google Scholar] [CrossRef]
- Bizet, A.A.; Liu, K.; Tran-Khanh, N.; Saksena, A.; Vorstenbosch, J.; Finnson, K.W.; Buschmann, M.D.; Philip, A. The tgf-beta co-receptor, cd109, promotes internalization and degradation of tgf-beta receptors. Biochim. Biophys. Acta 2011, 1813, 742–753. [Google Scholar] [CrossRef]
- Chen, C.L.; Liu, I.H.; Fliesler, S.J.; Han, X.; Huang, S.S.; Huang, J.S. Cholesterol suppresses cellular tgf-beta responsiveness: Implications in atherogenesis. J. Cell Sci. 2007, 120, 3509–3521. [Google Scholar] [CrossRef]
- Chen, C.L.; Wu, D.C.; Liu, M.Y.; Lin, M.W.; Huang, H.T.; Huang, Y.B.; Chen, L.C.; Chen, Y.Y.; Chen, J.J.; Yang, P.H.; et al. Cholest-4-en-3-one attenuates tgf-beta responsiveness by inducing tgf-beta receptors degradation in mv1lu cells and colorectal adenocarcinoma cells. J. Recept. Signal. Transduct. Res. 2017, 37, 189–199. [Google Scholar] [CrossRef]
- Chen, C.L.; Chen, C.Y.; Chen, Y.P.; Huang, Y.B.; Lin, M.W.; Wu, D.C.; Huang, H.T.; Liu, M.Y.; Chang, H.W.; Kao, Y.C.; et al. Betulinic acid enhances tgf-beta signaling by altering tgf-beta receptors partitioning between lipid-raft/caveolae and non-caveolae membrane microdomains in mink lung epithelial cells. J. Biomed. Sci. 2016, 23, 30. [Google Scholar] [CrossRef]
- Chen, C.L.; Chen, Y.P.; Lin, M.W.; Huang, Y.B.; Chang, F.R.; Duh, T.H.; Wu, D.C.; Wu, W.C.; Kao, Y.C.; Yang, P.H. Euphol from euphorbia tirucalli negatively modulates tgf-beta responsiveness via tgf-beta receptor segregation inside membrane rafts. PLoS ONE 2015, 10, e0140249. [Google Scholar]
- Miaczynska, M.; Pelkmans, L.; Zerial, M. Not just a sink: Endosomes in control of signal transduction. Curr. Opin. Cell Biol. 2004, 16, 400–406. [Google Scholar] [CrossRef]
- Huang, S.S.; Huang, J.S. Tgf-beta control of cell proliferation. J. Cell Biochem. 2005, 96, 447–462. [Google Scholar] [CrossRef]
- Chen, Y.G. Endocytic regulation of tgf-beta signaling. Cell Res. 2009, 19, 58–70. [Google Scholar] [CrossRef]
- Yakymovych, I.; Yakymovych, M.; Zang, G.; Mu, Y.; Bergh, A.; Landstrom, M.; Heldin, C.H. Cin85 modulates tgfbeta signaling by promoting the presentation of tgfbeta receptors on the cell surface. J. Cell Biol. 2015, 210, 319–332. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, L.; Luo, X.; Zhang, Y.; Ding, Q.; Jiang, X.; Wang, X.; Pan, Y.; Chen, Y. Tollip, an intracellular trafficking protein, is a novel modulator of the transforming growth factor-beta signaling pathway. J. Biol. Chem. 2012, 287, 39653–39663. [Google Scholar] [CrossRef]
- Capocasale, R.J.; Lamb, R.J.; Vonderheid, E.C.; Fox, F.E.; Rook, A.H.; Nowell, P.C.; Moore, J.S. Reduced surface expression of transforming growth factor beta receptor type ii in mitogen-activated t cells from sezary patients. Proc. Natl. Acad. Sci. USA 1995, 92, 5501–5505. [Google Scholar] [CrossRef]
- Qiu, Q.; Su, Y.; Zheng, Y.; Cai, H.; Wu, S.; Lu, W.; Zheng, W.; Shu, X.O.; Cai, Q. Increased psmad2 expression and cytoplasmic predominant presence of tgf-betarii in breast cancer tissue are associated with poor prognosis: Results from the shanghai breast cancer study. Breast Cancer Res. Treat. 2015, 149, 467–477. [Google Scholar] [CrossRef]
- Park, I.; Son, H.K.; Che, Z.M.; Kim, J. A novel gain-of-function mutation of tgf-beta receptor ii promotes cancer progression via delayed receptor internalization in oral squamous cell carcinoma. Cancer Lett 2012, 315, 161–169. [Google Scholar] [CrossRef]
- Chung, C.L.; Wang, S.W.; Martin, R.; Knolker, H.J.; Kao, Y.C.; Lin, M.H.; Chen, J.J.; Huang, Y.B.; Wu, D.C.; Chen, C.L. Pentachloropseudilin inhibits transforming growth factor-beta (tgf-beta) activity by accelerating cell-surface type ii tgf-beta receptor turnover in target cells. Chembiochem 2018, 19, 851–864. [Google Scholar] [CrossRef]
- Shih-Wei, W.; Chih-Ling, C.; Kao, Y.C.; Martin, R.; Knolker, H.J.; Shiao, M.S.; Chen, C.L. Pentabromopseudilin: A myosin v inhibitor suppresses tgf-beta activity by recruiting the type ii tgf-beta receptor to lysosomal degradation. J. Enzym. Inhib. Med. Chem. 2018, 33, 920–935. [Google Scholar] [CrossRef]
- Maravillas-Montero, J.L.; Santos-Argumedo, L. The myosin family: Unconventional roles of actin-dependent molecular motors in immune cells. J. Leukoc. Biol. 2012, 91, 35–46. [Google Scholar] [CrossRef]
- Liu, K.C.; Cheney, R.E. Myosins in cell junctions. Bioarchitecture 2012, 2, 158–170. [Google Scholar] [CrossRef]
- Cheney, R.E.; Mooseker, M.S. Unconventional myosins. Curr. Opin. Cell Biol. 1992, 4, 27–35. [Google Scholar] [CrossRef]
- Doberstein, S.K.; Pollard, T.D. Localization and specificity of the phospholipid and actin binding sites on the tail of acanthamoeba myosin ic. J. Cell. Biol. 1992, 117, 1241–1249. [Google Scholar] [CrossRef]
- Mayer, B.J. Sh3 domains: Complexity in moderation. J. Cell Sci. 2001, 114, 1253–1263. [Google Scholar]
- Nowak, G.; Pestic-Dragovich, L.; Hozak, P.; Philimonenko, A.; Simerly, C.; Schatten, G.; de Lanerolle, P. Evidence for the presence of myosin i in the nucleus. J. Biol. Chem. 1997, 272, 17176–17181. [Google Scholar] [CrossRef]
- Hofmann, W.A.; Vargas, G.M.; Ramchandran, R.; Stojiljkovic, L.; Goodrich, J.A.; de Lanerolle, P. Nuclear myosin i is necessary for the formation of the first phosphodiester bond during transcription initiation by rna polymerase ii. J. Cell Biochem. 2006, 99, 1001–1009. [Google Scholar] [CrossRef]
- Arif, E.; Solanki, A.K.; Srivastava, P.; Rahman, B.; Tash, B.R.; Holzman, L.B.; Janech, M.G.; Martin, R.; Knolker, H.J.; Fitzgibbon, W.R.; et al. The motor protein myo1c regulates transforming growth factor-beta-signaling and fibrosis in podocytes. Kidney Int. 2019, 96, 139–158. [Google Scholar] [CrossRef]
- Brandstaetter, H.; Kishi-Itakura, C.; Tumbarello, D.A.; Manstein, D.J.; Buss, F. Loss of functional myo1c/myosin 1c, a motor protein involved in lipid raft trafficking, disrupts autophagosome-lysosome fusion. Autophagy 2014, 10, 2310–2323. [Google Scholar] [CrossRef]
- Brandstaetter, H.; Kendrick-Jones, J.; Buss, F. Myo1c regulates lipid raft recycling to control cell spreading, migration and salmonella invasion. J. Cell Sci. 2012, 125, 1991–2003. [Google Scholar] [CrossRef]
- Hokanson, D.E.; Laakso, J.M.; Lin, T.; Sept, D.; Ostap, E.M. Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol. Biol. Cell 2006, 17, 4856–4865. [Google Scholar] [CrossRef]
- Golub, T.; Caroni, P. Pi(4,5)p2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. J. Cell Biol. 2005, 169, 151–165. [Google Scholar] [CrossRef]
- Barile, M.; Pisitkun, T.; Yu, M.J.; Chou, C.L.; Verbalis, M.J.; Shen, R.F.; Knepper, M.A. Large scale protein identification in intracellular aquaporin-2 vesicles from renal inner medullary collecting duct. Mol. Cell Proteom. 2005, 4, 1095–1106. [Google Scholar] [CrossRef]
- Chen, X.W.; Leto, D.; Chiang, S.H.; Wang, Q.; Saltiel, A.R. Activation of rala is required for insulin-stimulated glut4 trafficking to the plasma membrane via the exocyst and the motor protein myo1c. Dev. Cell 2007, 13, 391–404. [Google Scholar] [CrossRef]
- Arif, E.; Wagner, M.C.; Johnstone, D.B.; Wong, H.N.; George, B.; Pruthi, P.A.; Lazzara, M.J.; Nihalani, D. Motor protein myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein neph1 to the podocyte membrane. Mol. Cell Biol. 2011, 31, 2134–2150. [Google Scholar] [CrossRef]
- Krendel, M.; Osterweil, E.K.; Mooseker, M.S. Myosin 1e interacts with synaptojanin-1 and dynamin and is involved in endocytosis. Febs Lett 2007, 581, 644–650. [Google Scholar] [CrossRef]
- Cheng, J.; Grassart, A.; Drubin, D.G. Myosin 1e coordinates actin assembly and cargo trafficking during clathrin-mediated endocytosis. Mol. Biol. Cell 2012, 23, 2891–2904. [Google Scholar] [CrossRef]
- Hallett, R.M.; Dvorkin-Gheva, A.; Bane, A.; Hassell, J.A. A gene signature for predicting outcome in patients with basal-like breast cancer. Sci. Rep. 2012, 2, 227. [Google Scholar] [CrossRef]
- Ouderkirk-Pecone, J.L.; Goreczny, G.J.; Chase, S.E.; Tatum, A.H.; Turner, C.E.; Krendel, M. Myosin 1e promotes breast cancer malignancy by enhancing tumor cell proliferation and stimulating tumor cell de-differentiation. Oncotarget 2016, 7, 46419–46432. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ortega, O.; Santos-Argumedo, L. Myosin 1g contributes to cd44 adhesion protein and lipid rafts recycling and controls cd44 capping and cell migration in b lymphocytes. Front. Immunol. 2017, 8, 1731. [Google Scholar] [CrossRef]
- Patino-Lopez, G.; Aravind, L.; Dong, X.; Kruhlak, M.J.; Ostap, E.M.; Shaw, S. Myosin 1g is an abundant class i myosin in lymphocytes whose localization at the plasma membrane depends on its ancient divergent pleckstrin homology (ph) domain (myo1ph). J. Biol. Chem. 2010, 285, 8675–8686. [Google Scholar] [CrossRef]
- Maravillas-Montero, J.L.; Lopez-Ortega, O.; Patino-Lopez, G.; Santos-Argumedo, L. Myosin 1g regulates cytoskeleton plasticity, cell migration, exocytosis, and endocytosis in b lymphocytes. Eurj. Immunol. 2014, 44, 877–886. [Google Scholar] [CrossRef]
- Buschow, S.I.; van Balkom, B.W.; Aalberts, M.; Heck, A.J.; Wauben, M.; Stoorvogel, W. Mhc class ii-associated proteins in b-cell exosomes and potential functional implications for exosome biogenesis. Immunol. Cell Biol. 2010, 88, 851–856. [Google Scholar] [CrossRef]
- Linkermann, A.; Gelhaus, C.; Lettau, M.; Qian, J.; Kabelitz, D.; Janssen, O. Identification of interaction partners for individual sh3 domains of fas ligand associated members of the pch protein family in t lymphocytes. Biochim. Biophys. Acta 2009, 1794, 168–176. [Google Scholar] [CrossRef]
- Zhang, W.B.; Yao, L.L.; Li, X.D. The globular tail domain of myosin-5a functions as a dimer in regulating the motor activity. J. Biol. Chem. 2016, 291, 13571–13579. [Google Scholar] [CrossRef]
- Sellers, J.R.; Thirumurugan, K.; Sakamoto, T.; Hammer, J.A., 3rd; Knight, P.J. Calcium and cargoes as regulators of myosin 5a activity. Biochem. Biophys. Res. Commun. 2008, 369, 176–181. [Google Scholar] [CrossRef]
- Rogers, S.L.; Gelfand, V.I. Myosin cooperates with microtubule motors during organelle transport in melanophores. Curr. Biol. 1998, 8, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.L.; Lee, A.J.; Bridgman, P.C.; Mooseker, M.S. Vesicle-associated brain myosin-v can be activated to catalyze actin-based transport. J. Cell Sci. 1998, 111, 2055–2066. [Google Scholar]
- Tabb, J.S.; Molyneaux, B.J.; Cohen, D.L.; Kuznetsov, S.A.; Langford, G.M. Transport of er vesicles on actin filaments in neurons by myosin v. J. Cell Sci. 1998, 111, 3221–3234. [Google Scholar]
- Woolner, S.; Bement, W.M. Unconventional myosins acting unconventionally. Trends Cell Biol. 2009, 19, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.T.; Chang, W.; Masters, S.E.; Mooseker, M.S. Myosin-va binds to and mechanochemically couples microtubules to actin filaments. Mol. Biol. Cell 2004, 15, 151–161. [Google Scholar] [CrossRef]
- Lise, M.F.; Wong, T.P.; Trinh, A.; Hines, R.M.; Liu, L.; Kang, R.; Hines, D.J.; Lu, J.; Goldenring, J.R.; Wang, Y.T.; et al. Involvement of myosin vb in glutamate receptor trafficking. J. Biol. Chem. 2006, 281, 3669–3678. [Google Scholar] [CrossRef]
- Millman, E.E.; Zhang, H.; Zhang, H.; Godines, V.; Bean, A.J.; Knoll, B.J.; Moore, R.H. Rapid recycling of beta-adrenergic receptors is dependent on the actin cytoskeleton and myosin vb. Traffic 2008, 9, 1958–1971. [Google Scholar] [CrossRef]
- Yan, Q.; Sun, W.; Kujala, P.; Lotfi, Y.; Vida, T.A.; Bean, A.J. Cart: An hrs/actinin-4/berp/myosin v protein complex required for efficient receptor recycling. Mol. Biol. Cell 2005, 16, 2470–2482. [Google Scholar] [CrossRef]
- Buss, F.; Kendrick-Jones, J. How are the cellular functions of myosin vi regulated within the cell? Biochem Biophys Res. Commun. 2008, 369, 165–175. [Google Scholar] [CrossRef]
- Penheiter, S.G.; Singh, R.D.; Repellin, C.E.; Wilkes, M.C.; Edens, M.; Howe, P.H.; Pagano, R.E.; Leof, E.B. Type ii transforming growth factor-beta receptor recycling is dependent upon the clathrin adaptor protein dab2. Mol. Biol. Cell 2010, 21, 4009–4019. [Google Scholar] [CrossRef]
- Homma, K.; Saito, J.; Ikebe, R.; Ikebe, M. Motor function and regulation of myosin x. J. Biol Chem 2001, 276, 34348–34354. [Google Scholar] [CrossRef]
- Berg, J.S.; Derfler, B.H.; Pennisi, C.M.; Corey, D.P.; Cheney, R.E. Myosin-x, a novel myosin with pleckstrin homology domains, associates with regions of dynamic actin. J. Cell Sci. 2000, 113, 3439–3451. [Google Scholar]
- Rechsteiner, M.; Rogers, S.W. Pest sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar] [CrossRef]
- Dvornikov, D.; Schneider, M.A.; Ohse, S.; Szczygiel, M.; Titkova, I.; Rosenblatt, M.; Muley, T.; Warth, A.; Herth, F.J.; Dienemann, H.; et al. Expression ratio of the tgfbeta-inducible gene myo10 is prognostic for overall survival of squamous cell lung cancer patients and predicts chemotherapy response. Sci. Rep. 2018, 8, 9517. [Google Scholar] [CrossRef]
- Sousa, A.D.; Cheney, R.E. Myosin-x: A molecular motor at the cell’s fingertips. Trends Cell Biol. 2005, 15, 533–539. [Google Scholar] [CrossRef]
- Arjonen, A.; Kaukonen, R.; Mattila, E.; Rouhi, P.; Hognas, G.; Sihto, H.; Miller, B.W.; Morton, J.P.; Bucher, E.; Taimen, P.; et al. Mutant p53-associated myosin-x upregulation promotes breast cancer invasion and metastasis. J. Clin. Invest. 2014, 124, 1069–1082. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, C.-L.; Tai, S.-B.; Hu, T.-H.; Chen, J.-J.; Chen, C.-L. Roles of Myosin-Mediated Membrane Trafficking in TGF-β Signaling. Int. J. Mol. Sci. 2019, 20, 3913. https://doi.org/10.3390/ijms20163913
Chung C-L, Tai S-B, Hu T-H, Chen J-J, Chen C-L. Roles of Myosin-Mediated Membrane Trafficking in TGF-β Signaling. International Journal of Molecular Sciences. 2019; 20(16):3913. https://doi.org/10.3390/ijms20163913
Chicago/Turabian StyleChung, Chih-Ling, Shun-Ban Tai, Tsung-Hui Hu, Jih-Jung Chen, and Chun-Lin Chen. 2019. "Roles of Myosin-Mediated Membrane Trafficking in TGF-β Signaling" International Journal of Molecular Sciences 20, no. 16: 3913. https://doi.org/10.3390/ijms20163913
APA StyleChung, C. -L., Tai, S. -B., Hu, T. -H., Chen, J. -J., & Chen, C. -L. (2019). Roles of Myosin-Mediated Membrane Trafficking in TGF-β Signaling. International Journal of Molecular Sciences, 20(16), 3913. https://doi.org/10.3390/ijms20163913