Combination of Fetal Fraction Estimators Based on Fragment Lengths and Fragment Counts in Non-Invasive Prenatal Testing

 , , ,
, , ,
Abstract
:1. Introduction
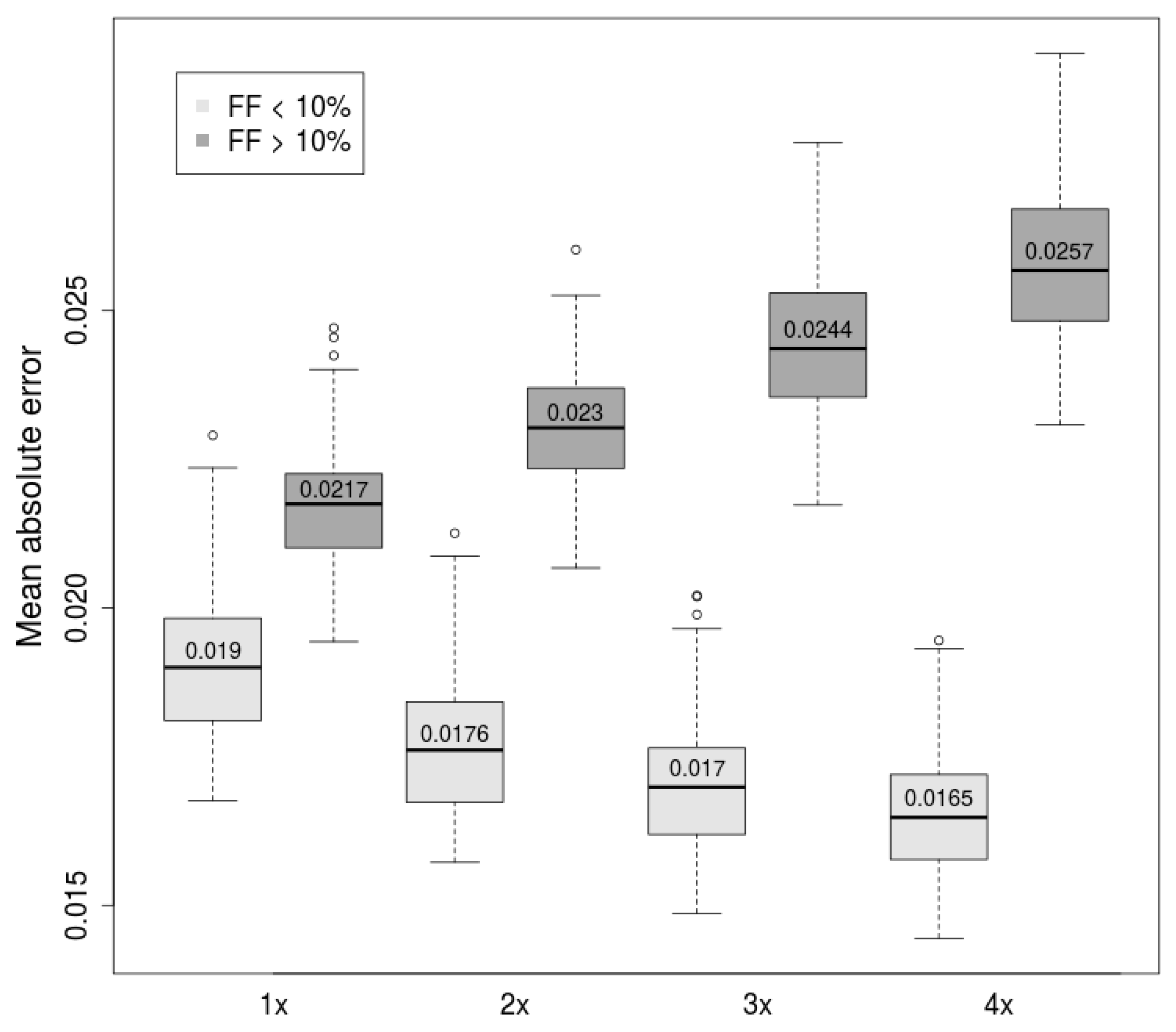
2. Results
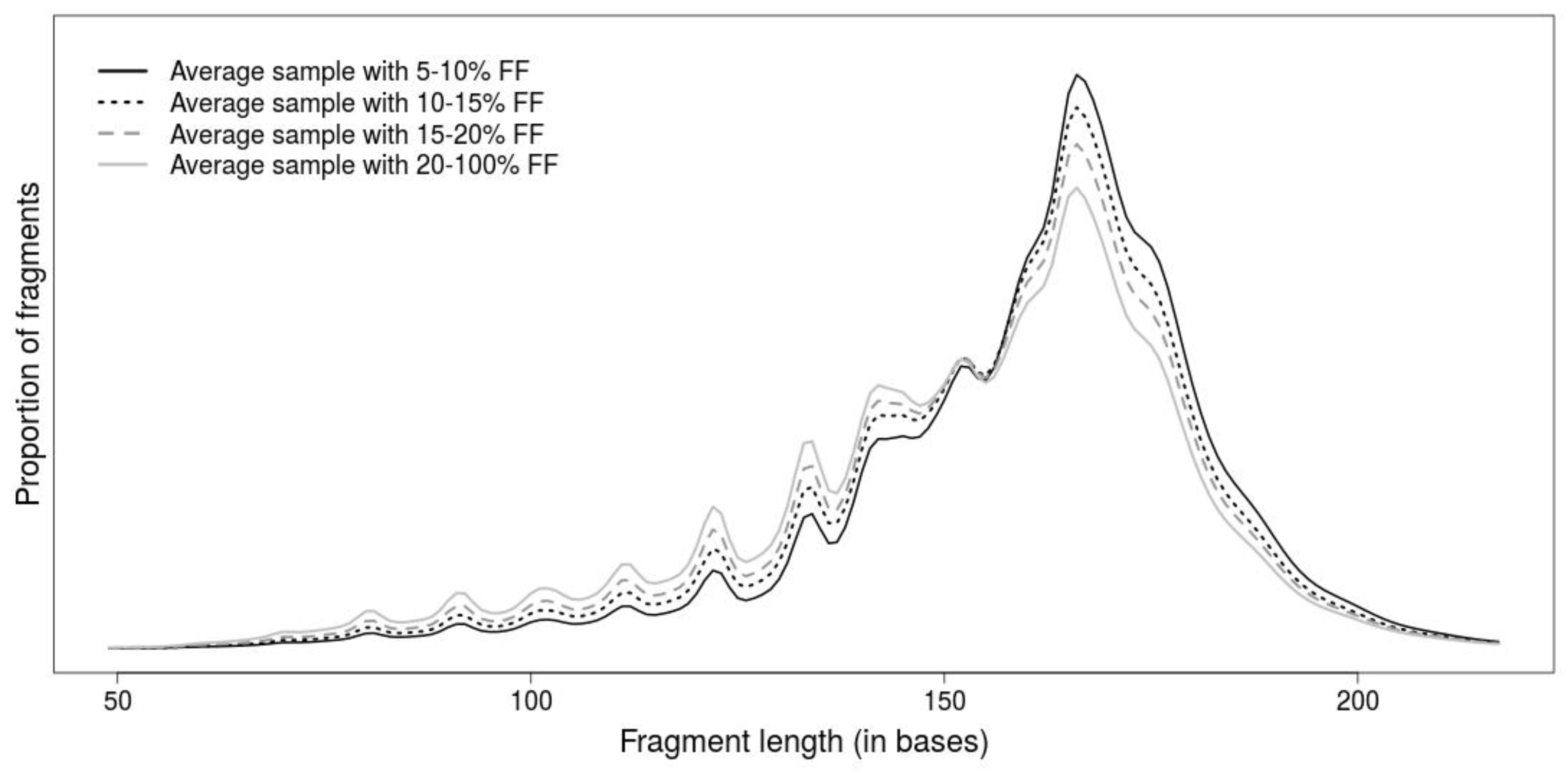
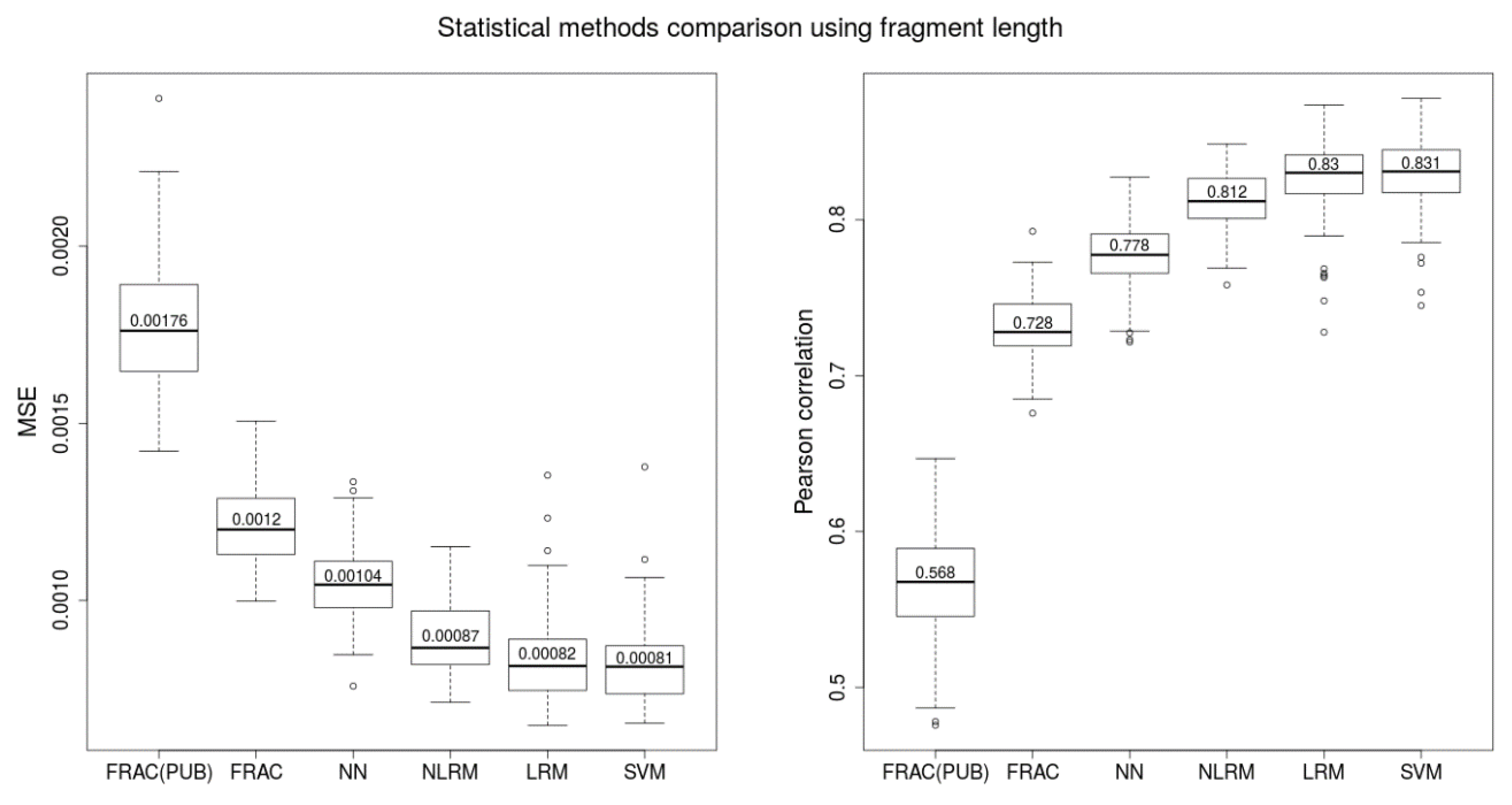
2.1. Comparison of Length-Based Methods
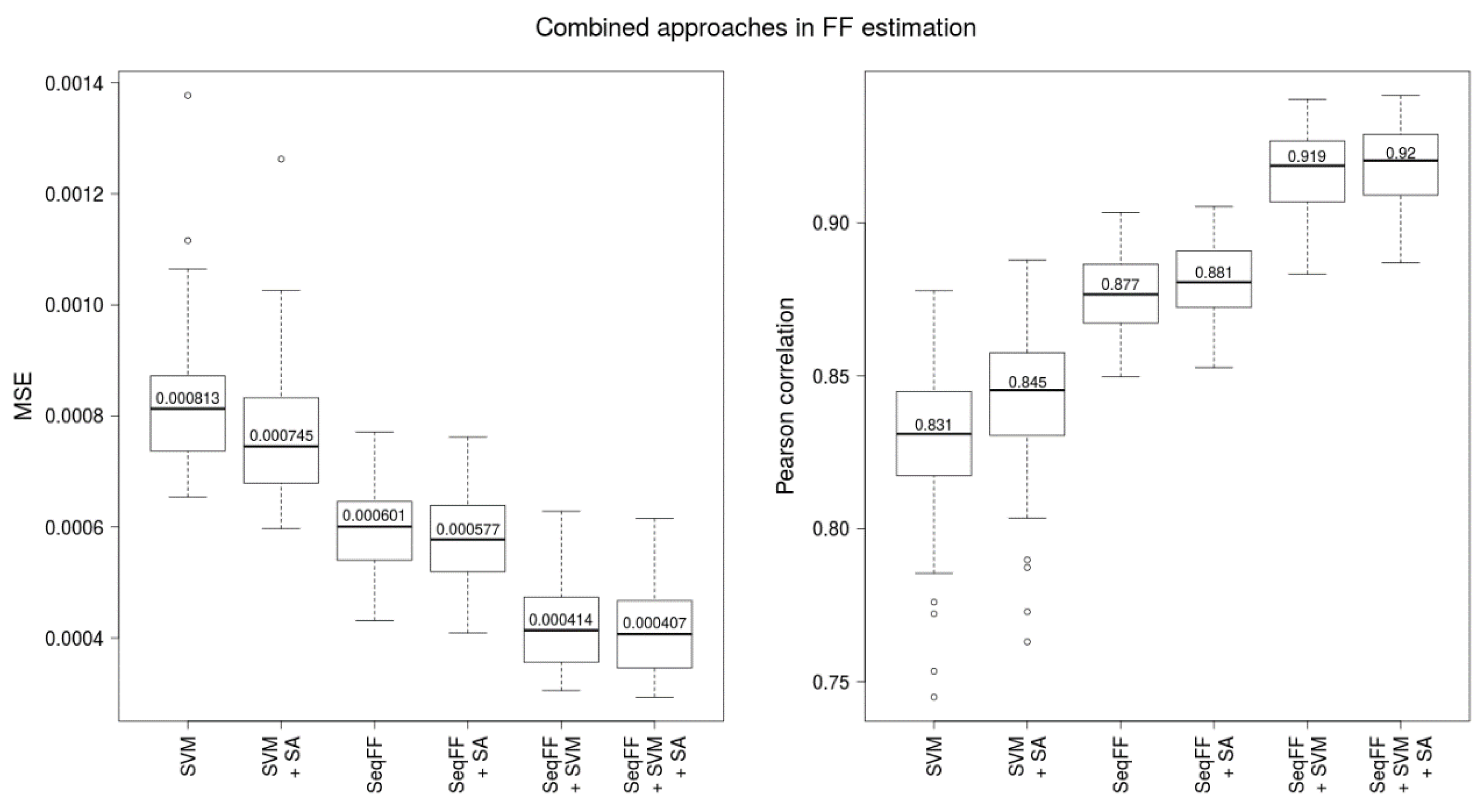
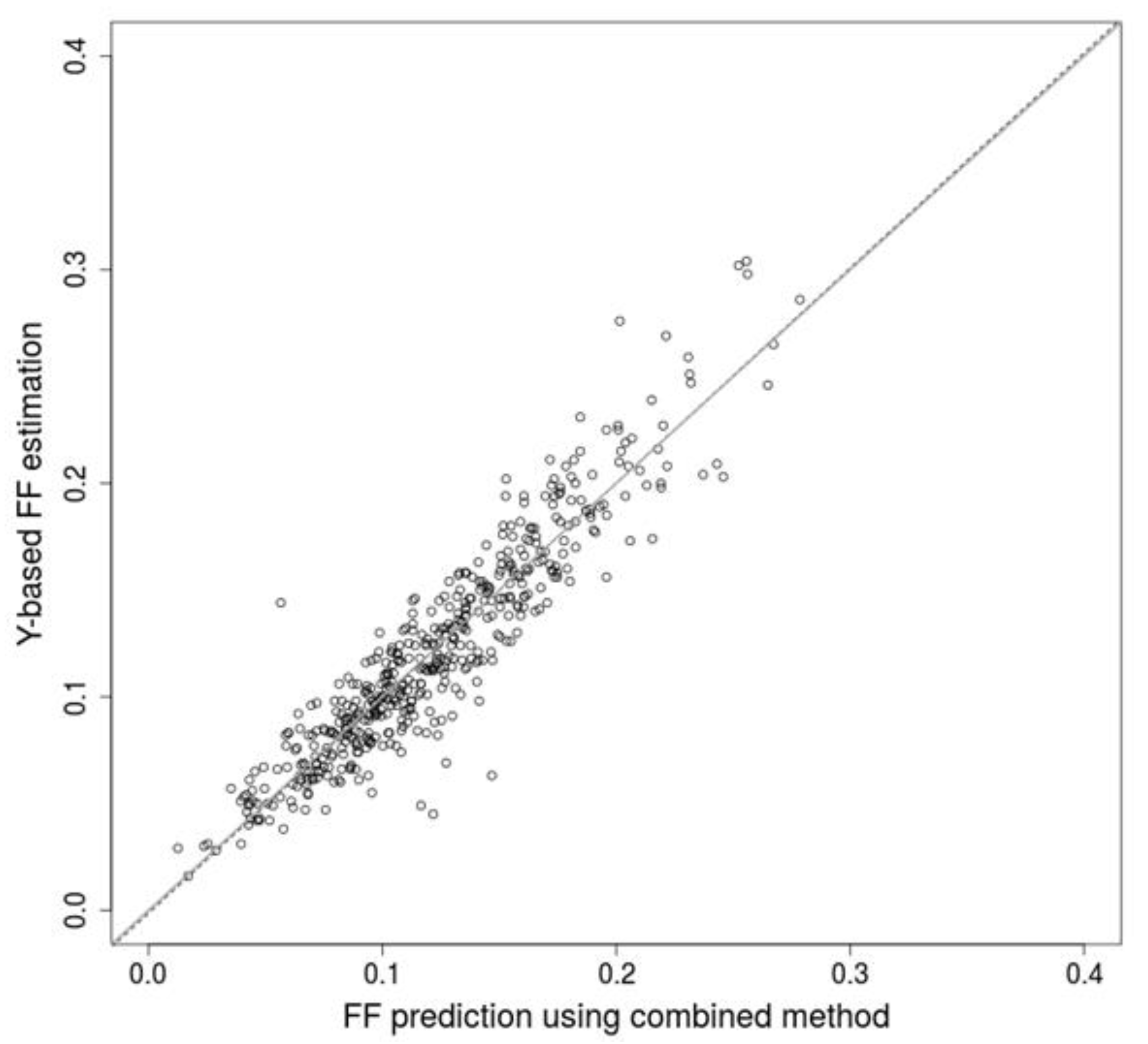
2.2. Combination of Methods Improves Prediction Accuracy
2.3. Weighting of Samples with Low Fraction
3. Discussion
4. Materials and Methods
4.1. Sample Acquisition
4.2. Sample Preparation and Sequencing
4.3. Mapping and GC Correction
4.4. Data Preparation and Evaluation of Models
Y-Based Estimator
4.5. FF Estimators Based on Fragment Length
4.5.1. Linear Regression Model (LRM)
4.5.2. Non-Linear Regression Model (NLRM)
4.5.3. Neural Network (NN)
4.5.4. Support Vector Machine (SVM)
4.6. FF Estimators Based on Read Counts
SeqFF
4.7. Correlation with Sample Attributes
4.8. Combinaton of FF Estimators
4.9. Weighted Samples
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NIPT | non-invasive prenatal testing |
| FF | fetal fraction |
| NN | neural network |
| SVM | support vector machine |
| cfDNA | cell free DNA |
| cffDNA | cell free fetal DNA |
| LRM | linear regression model |
| NLRM | non-linear regression model |
References
- Pös, O.; Biró, O.; Szemes, T.; Nagy, B. Circulating cell-free nucleic acids: Characteristics and applications. Eur. J. Hum. Genet. 2018, 26, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Allyse, M.; Minear, M.A.; Berson, E.; Sridhar, S.; Rote, M.; Hung, A.; Chandrasekharan, S. Non-invasive prenatal testing: A review of international implementation and challenges. Int. J. Womens Health 2015, 7, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.D.; Dennis Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.G.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Budiš, J.; Kucharík, M.; Duriš, F.; Gazdarica, J.; Zrubcová, M.; Ficek, A.; Szemes, T.; Brejová, B.; Radvanszky, J. Dante: Genotyping of known complex and expanded short tandem repeats. Bioinformatics 2018, 35, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Alfirevic, Z.; Mujezinovic, F.; Sundberg, K. Amniocentesis and chorionic villus sampling for prenatal diagnosis. Cochrane Database Syst. Rev. 2003. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Lamar Parker, R.; Wentworth, J.; Madankumar, R.; Saffer, C.; Das, A.F.; Craig, J.A.; Chudova, D.I.; Devers, P.L.; Jones, K.W.; et al. DNA Sequencing Versus Standard Prenatal Aneuploidy Screening. Obstet. Gynecol. Surv. 2014, 69, 319–321. [Google Scholar] [CrossRef]
- Ashoor, G.; Syngelaki, A.; Poon, L.C.Y.; Rezende, J.C.; Nicolaides, K.H. Fetal fraction in maternal plasma cell-free DNA at 11–13 weeks’ gestation: Relation to maternal and fetal characteristics. Ultrasound Obstet. Gynecol. 2013, 41, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; van den Boom, D.; Bombard, A.T.; Deciu, C.; Grody, W.W.; et al. DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet. Med. 2011, 13, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Canick, J.A.; Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E. The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat. Diagn. 2013, 33, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Rava, R.P.; Srinivasan, A.; Sehnert, A.J.; Bianchi, D.W. Circulating Fetal Cell-Free DNA Fractions Differ in Autosomal Aneuploidies and Monosomy X. Clin. Chem. 2013, 60, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.L.; Jiang, P. Bioinformatics Approaches for Fetal DNA Fraction Estimation in Noninvasive Prenatal Testing. Int. J. Mol. Sci. 2017, 18, 453. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, R.W.K.; Akolekar, R.; Zheng, Y.W.L.; Leung, T.Y.; Sun, H.; Chan, K.C.A.; Lun, F.M.F.; Go, A.T.J.I.; Lau, E.T.; To, W.W.K.; et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: Large scale validity study. BMJ 2011, 342, c7401. [Google Scholar] [CrossRef] [PubMed]
- Duris, F.; Gazdarica, J.; Gazdaricova, I.; Strieskova, L.; Budis, J.; Turna, J.; Szemes, T. Mean and variance of ratios of proportions from categories of a multinomial distribution. J. Stat. Distrib. Appl. 2018, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Chan, K.C.A.; Liao, G.J.W.; Zheng, Y.W.L.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D.; Sun, H. FetalQuant: Deducing fractional fetal DNA concentration from massively parallel sequencing of DNA in maternal plasma. Bioinformatics 2012, 28, 2883–2890. [Google Scholar] [CrossRef]
- Jiang, P.; Peng, X.; Su, X.; Sun, K.; Yu, S.C.Y.; Chu, W.I.; Leung, T.Y.; Sun, H.; Chiu, R.W.K.; Lo, Y.M.D.; et al. FetalQuantSD: Accurate quantification of fetal DNA fraction by shallow-depth sequencing of maternal plasma DNA. NPJ Genom. Med. 2016, 1, 16103. [Google Scholar] [CrossRef] [PubMed]
- Straver, R.; Oudejans, C.B.M.; Sistermans, E.A.; Reinders, M.J.T. Calculating the fetal fraction for noninvasive prenatal testing based on genome-wide nucleosome profiles. Prenat. Diagn. 2016, 36, 614–621. [Google Scholar] [CrossRef]
- Kim, S.K.; Hannum, G.; Geis, J.; Tynan, J.; Hogg, G.; Zhao, C.; Jensen, T.J.; Mazloom, A.R.; Oeth, P.; Ehrich, M.; et al. Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenat. Diagn. 2015, 35, 810–815. [Google Scholar] [CrossRef]
- van Beek, D.M.; Straver, R.; Weiss, M.M.; Boon, E.M.J.; Huijsdens-van Amsterdam, K.; Oudejans, C.B.M.; Reinders, M.J.T.; Sistermans, E.A. Comparing methods for fetal fraction determination and quality control of NIPT samples. Prenat. Diagn. 2017, 37, 769–773. [Google Scholar] [CrossRef]
- Minarik, G.; Repiska, G.; Hyblova, M.; Nagyova, E.; Soltys, K.; Budis, J.; Duris, F.; Sysak, R.; Gerykova Bujalkova, M.; Vlkova-Izrael, B.; et al. Utilization of Benchtop Next Generation Sequencing Platforms Ion Torrent PGM and MiSeq in Noninvasive Prenatal Testing for Chromosome 21 Trisomy and Testing of Impact of In Silico and Physical Size Selection on Its Analytical Performance. PLoS ONE 2015, 10, e0144811. [Google Scholar] [CrossRef]
- Budis, J.; Gazdarica, J.; Radvanszky, J.; Szucs, G.; Kucharik, M.; Strieskova, L.; Gazdaricova, I.; Harsanyova, M.; Duris, F.; Minarik, G.; et al. Innovative method for reducing uninformative calls in non-invasive prenatal testing. arXiv 2018, arXiv:1806.08552. [Google Scholar]
- Shubina, J.; Trofimov, D.Y.; Barkov, I.Y.; Stupko, O.K.; Goltsov, A.Y.; Mukosey, I.S.; Tetruashvili, N.K.; Kim, L.V.; Bakharev, V.A.; Karetnikova, N.A.; et al. In silico size selection is effective in reducing false positive NIPS cases of monosomy X that are due to maternal mosaic monosomy X. Prenat. Diagn. 2017, 37, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.C.Y.; Chan, K.C.A.; Zheng, Y.W.L.; Jiang, P.; Liao, G.J.W.; Sun, H.; Akolekar, R.; Leung, T.Y.; Go, A.T.J.I.; van Vugt, J.M.G.; et al. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA 2014, 111, 8583–8588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.J.; Figueiredo, M.A.T. Boosting Algorithms: A Review of Methods, Theory, and Applications. In Ensemble Machine Learning; Springer: Boston, MA, USA, 2012; pp. 35–85. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Yin, A.-H.; Peng, C.-F.; Fu, F.; Yang, J.-X.; Li, R.; Chen, Y.-Y.; Luo, D.-H.; Zhang, Y.-L.; Ou, Y.-M.; et al. Noninvasive prenatal diagnosis of common aneuploidies by semiconductor sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7415–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudecova, I.; Sahota, D.; Heung, M.M.S.; Jin, Y.; Lee, W.S.; Leung, T.Y.; Lo, Y.M.D.; Chiu, R.W.K. Maternal Plasma Fetal DNA Fractions in Pregnancies with Low and High Risks for Fetal Chromosomal Aneuploidies. PLoS ONE 2014, 9, e88484. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Byrd, R.H.; Lu, P.; Nocedal, J. Algorithm 778: L-BFGS-B: Fortran subroutines for large-scale bound-constrained optimization. ACM Trans. Math. Softw. 1997, 23, 550–560. [Google Scholar] [CrossRef]
- Garreta, R.; Moncecchi, G. Learning Scikit-Learn: Machine Learning in Python; Packt Publishing Ltd.: Birmingham, UK, 2013; ISBN 9781783281947. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | SeqFF + SA | SVM + SA | SVM + SeqFF | Seqff + SVM + SA |
|---|---|---|---|---|
| SVM | – | 0.0418 | 0.0269 | 0.0243 |
| SeqFF | 1.1325 | – | 0.0237 | 0.0255 |
| BMI | −0.0006 | −0.0058 | – | −0.0019 |
| LC | −0.0020 | −0.0005 | – | −0.0015 |
| GA | ~0.0000 | 0.0037 | – | 0.0016 |
| Intercept | 0.0204 | 0.1222 | 0.1223 | 0.1227 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazdarica, J.; Hekel, R.; Budis, J.; Kucharik, M.; Duris, F.; Radvanszky, J.; Turna, J.; Szemes, T. Combination of Fetal Fraction Estimators Based on Fragment Lengths and Fragment Counts in Non-Invasive Prenatal Testing. Int. J. Mol. Sci. 2019, 20, 3959. https://doi.org/10.3390/ijms20163959
Gazdarica J, Hekel R, Budis J, Kucharik M, Duris F, Radvanszky J, Turna J, Szemes T. Combination of Fetal Fraction Estimators Based on Fragment Lengths and Fragment Counts in Non-Invasive Prenatal Testing. International Journal of Molecular Sciences. 2019; 20(16):3959. https://doi.org/10.3390/ijms20163959
Chicago/Turabian StyleGazdarica, Juraj, Rastislav Hekel, Jaroslav Budis, Marcel Kucharik, Frantisek Duris, Jan Radvanszky, Jan Turna, and Tomas Szemes. 2019. "Combination of Fetal Fraction Estimators Based on Fragment Lengths and Fragment Counts in Non-Invasive Prenatal Testing" International Journal of Molecular Sciences 20, no. 16: 3959. https://doi.org/10.3390/ijms20163959
APA StyleGazdarica, J., Hekel, R., Budis, J., Kucharik, M., Duris, F., Radvanszky, J., Turna, J., & Szemes, T. (2019). Combination of Fetal Fraction Estimators Based on Fragment Lengths and Fragment Counts in Non-Invasive Prenatal Testing. International Journal of Molecular Sciences, 20(16), 3959. https://doi.org/10.3390/ijms20163959