Targeting Cellular Metabolism Modulates Head and Neck Oncogenesis

 ,
,
Abstract
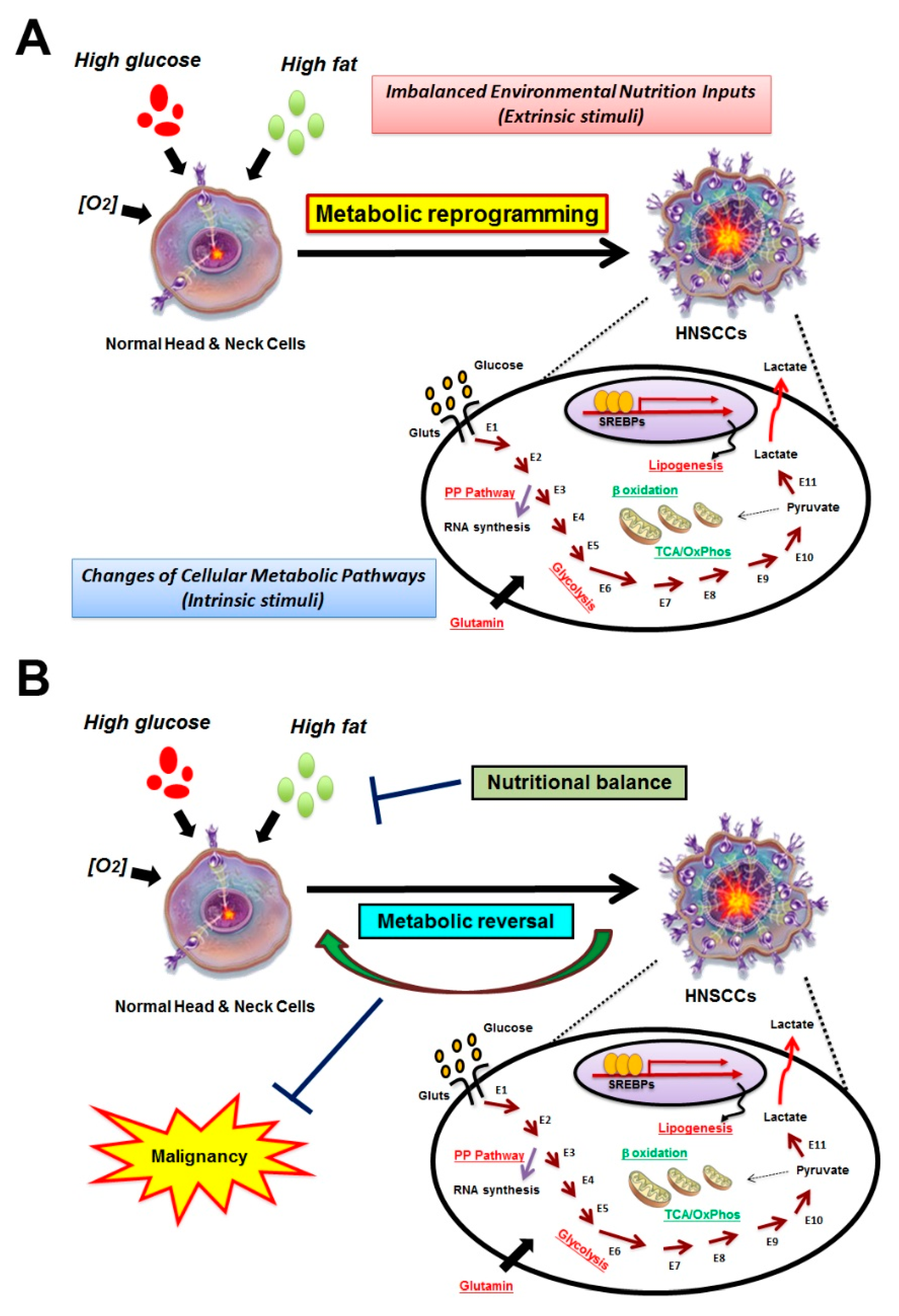
:1. Introduction
2. Identification for HNSCC-Specific Metabolic Profile
3. Association of Systemic Disease and HNSCCs
4. Development of Anti-Cancer Scheme by Targeting Distinct Metabolic Pathways in HNSCCs
4.1. Targeting Aerobic Glycolysis in HNSCCs
4.2. Targeting Mitochondrial Related Metabolism in HNSCCs
4.2.1. Reactive Oxygen Species (ROS) and Apoptotic Pathway
4.2.2. Tricarboxylic Acid Cycle (TCA Cycle) & Electron Transport Chain (ETC)
4.3. Targeting Lipid Metabolism in HNSCCs
4.4. Targeting Amino Acid Metabolism in HNSCCs
5. Molecular Basis of Metabolic Regulations in HNSCCs
5.1. Non-Coding RNA (ncRNA) Mediated Control for HNSCC Metabolic Cues
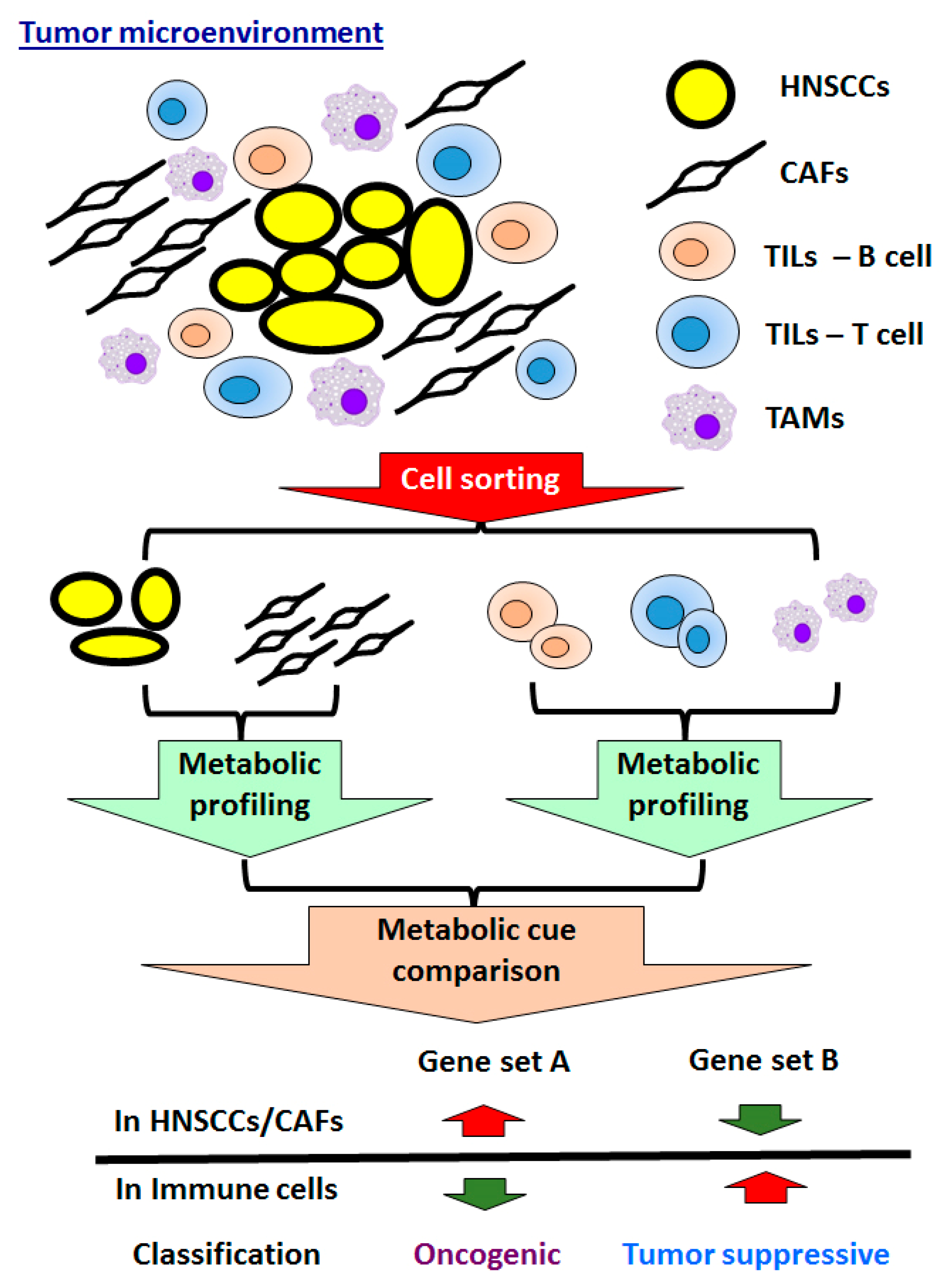
5.2. Crosstalk of Tumors and Microenvironment in HNSCCs
5.2.1. CAFs in HNSCCs
5.2.2. Immune Cells in HNSCCs
5.2.3. Hypoxia/Vascular Signals in HNSCCs
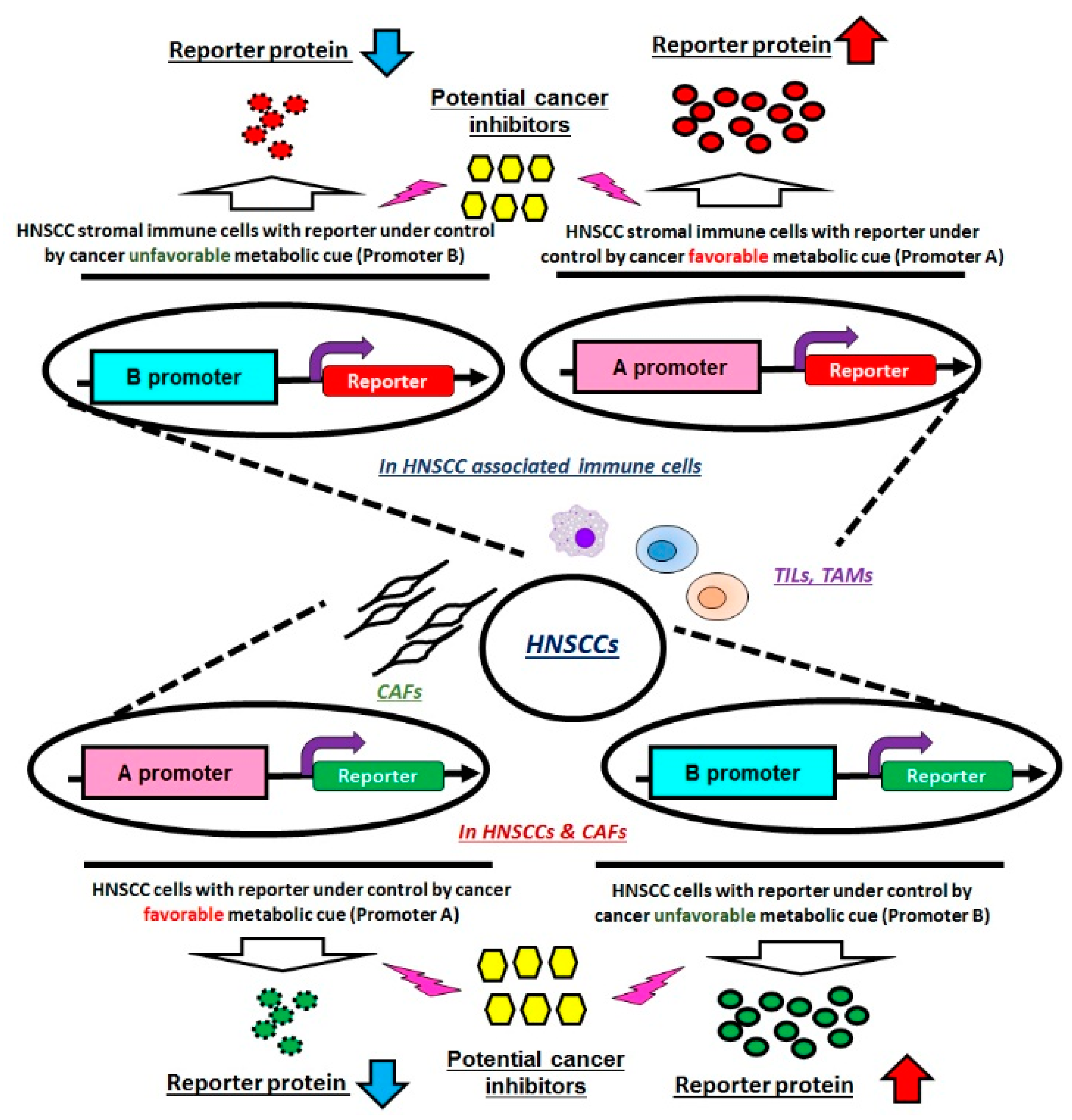
6. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Cohen, N.; Fedewa, S.; Chen, A.Y. Epidemiology and demographics of the head and neck cancer population. Oral Maxillofac Surg Clin. North. Am. 2018, 30, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Meurman, J.H. Infectious and dietary risk factors of oral cancer. Oral Oncol. 2010, 46, 411–413. [Google Scholar] [CrossRef]
- Cognetti, D.M.; Weber, R.S.; Lai, S.Y. Head and neck cancer: An evolving treatment paradigm. Cancer 2008, 113, 1911–1932. [Google Scholar] [CrossRef] [PubMed]
- Du, E.; Mazul, A.L.; Farquhar, D.; Brennan, P.; Anantharaman, D.; Abedi-Ardekani, B.; Weissler, M.C.; Hayes, D.N.; Olshan, A.F.; Zevallos, J.P. Long-term survival in head and neck cancer: Impact of site, stage, smoking, and human papillomavirus status. Laryngoscope 2019. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Santuray, R.T.; Johnson, D.E.; Grandis, J.R. New therapies in head and neck cancer. Trends Cancer 2018, 4, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Mascitti, M.; Rubini, C.; De Michele, F.; Balercia, P.; Girotto, R.; Troiano, G.; Lo Muzio, L.; Santarelli, A. American joint committee on cancer staging system 7th edition versus 8th edition: Any improvement for patients with squamous cell carcinoma of the tongue? Oral Surg. Oral Med. Oral Pathol Oral Radiol. 2018, 126, 415–423. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.; Poff, A.M.; D’Agostino, D.P.; Mukherjee, P. Metabolic therapy: A new paradigm for managing malignant brain cancer. Cancer Lett. 2015, 356, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab 2010, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Kim, D. Cancer metabolism: Fueling more than just growth. Mol. Cells 2016, 39, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Racker, E. History of the pasteur effect and its pathobiology. Mol. Cell Biochem. 1974, 5, 17–23. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, 129–150. [Google Scholar] [CrossRef]
- Chen, T.Y.; Hsieh, Y.T.; Huang, J.M.; Liu, C.J.; Chuang, L.T.; Huang, P.C.; Kuo, T.Y.; Chia, H.Y.; Chou, C.Y.; Chang, C.W.; et al. Determination of pyruvate metabolic fates modulates head and neck tumorigenesis. Neoplasia 2019, 21, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Gates, R.E.; Rees, R.S. Altered vitamin a-binding proteins in carcinoma of the head and neck. Cancer 1985, 56, 2598–2604. [Google Scholar] [CrossRef]
- Kim, J.S.; Steck, P.A.; Gallick, G.E.; Lee, J.S.; Blick, M.; Hong, W.K.; Lotan, R. Suppression by retinoic acid of epidermal growth factor receptor autophosphorylation and glycosylation in cultured human head and neck squamous carcinoma cells. J. Natl. Cancer Inst. Monogr 1992, 101–110. [Google Scholar]
- Bier, H.; Bergler, W.; Mende, S.; Ganzer, U. Glutathione content and gamma-glutamyltranspeptidase activity in squamous cell head and neck cancer xenografts. Arch. Otorhinolaryngol 1988, 245, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Janot, F.; Massaad, L.; Ribrag, V.; de Waziers, I.; Beaune, P.H.; Luboinski, B.; Parise, O., Jr.; Gouyette, A.; Chabot, G.G. Principal xenobiotic-metabolizing enzyme systems in human head and neck squamous cell carcinoma. Carcinogenesis 1993, 14, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, C.B.; Roy, N. Meta-analysis of glutathione s-transferase m1 genotype and risk toward head and neck cancer. Head Neck 2006, 28, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Edstrom, S.; Westin, T.; Delle, U.; Lundholm, K. Cell cycle distribution and ornithine decarboxylase activity in head and neck cancer in response to enteral nutrition. Eur. J. Cancer Clin. Oncol. 1989, 25, 227–232. [Google Scholar] [CrossRef]
- Rydell, E.L.; Axelsson, K.L.; Olofsson, J.; Hellem, S. Protein kinase activities in neoplastic squamous epithelia and normal epithelia from the upper aero-digestive tract. Cancer Biochem. Biophys 1990, 11, 187–194. [Google Scholar]
- Pizzorno, G.; Chang, Y.M.; McGuire, J.J.; Bertino, J.R. Inherent resistance of human squamous carcinoma cell lines to methotrexate as a result of decreased polyglutamylation of this drug. Cancer Res. 1989, 49, 5275–5280. [Google Scholar]
- Caldani, C.; Thyss, A.; Schneider, M.; Milano, G.; Buray, L.; Demard, F. Orosomucoid:Prealbumin ratio--a marker of the host-tumor relationship in head and neck cancer. Eur. J. Cancer Clin. Oncol. 1988, 24, 653–657. [Google Scholar] [CrossRef]
- Frank, J.L.; Lawrence, W., Jr.; Banks, W.L., Jr.; McKinnon, J.G.; Chan, W.M.; Collins, J.M. Modulation of cell cycle kinetics in human cancer with total parenteral nutrition. Cancer 1992, 69, 1858–1864. [Google Scholar] [CrossRef]
- Byerley, L.O.; Heber, D.; Bergman, R.N.; Dubria, M.; Chi, J. Insulin action and metabolism in patients with head and neck cancer. Cancer 1991, 67, 2900–2906. [Google Scholar] [CrossRef]
- Joslin, E.P.; Lombard, H.L.; Burrows, R.E.; Manning, M.D. Diabetes and cancer. N Engl. J. Med. 1959, 260, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Banoczy, J.; Dinya, E.; Tamas, G., Jr. Occurrence of oral leukoplakia and lichen planus in diabetes mellitus. J. Oral Pathol. Med. 1992, 21, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, T.; Reichart, P.A.; Scheifele, C. Clinical risk factors of oral leukoplakia in a representative sample of the us population. Oral Oncol. 2004, 40, 158–163. [Google Scholar] [CrossRef]
- Meisel, P.; Dau, M.; Sumnig, W.; Holtfreter, B.; Houshmand, M.; Nauck, M.; Kocher, T. Association between glycemia, serum lipoproteins, and the risk of oral leukoplakia: The population-based study of health in pomerania (ship). Diabetes Care 2010, 33, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Atchison, E.A.; Gridley, G.; Carreon, J.D.; Leitzmann, M.F.; McGlynn, K.A. Risk of cancer in a large cohort of u.S. Veterans with diabetes. Int. J. Cancer 2011, 128, 635–643. [Google Scholar] [CrossRef]
- Wu, C.H.; Wu, T.Y.; Li, C.C.; Lui, M.T.; Chang, K.W.; Kao, S.Y. Impact of diabetes mellitus on the prognosis of patients with oral squamous cell carcinoma: A retrospective cohort study. Ann. Surg Oncol. 2010, 17, 2175–2183. [Google Scholar] [CrossRef]
- Tseng, C.H. Oral cancer in taiwan: Is diabetes a risk factor? Clin. Oral Investig. 2013, 17, 1357–1364. [Google Scholar] [CrossRef]
- Goutzanis, L.; Vairaktaris, E.; Yapijakis, C.; Kavantzas, N.; Nkenke, E.; Derka, S.; Vassiliou, S.; Acil, Y.; Kessler, P.; Stavrianeas, N.; et al. Diabetes may increase risk for oral cancer through the insulin receptor substrate-1 and focal adhesion kinase pathway. Oral Oncol. 2007, 43, 165–173. [Google Scholar] [CrossRef]
- Liu, C.J.; Chang, W.J.; Chen, C.Y.; Sun, F.J.; Cheng, H.W.; Chen, T.Y.; Lin, S.C.; Li, W.C. Dynamic cellular and molecular modulations of diabetes mediated head and neck carcinogenesis. Oncotarget 2015, 6, 29268–29284. [Google Scholar] [PubMed] [Green Version]
- Beloueche-Babari, M.; Box, C.; Arunan, V.; Parkes, H.G.; Valenti, M.; De Haven Brandon, A.; Jackson, L.E.; Eccles, S.A.; Leach, M.O. Acquired resistance to egfr tyrosine kinase inhibitors alters the metabolism of human head and neck squamous carcinoma cells and xenograft tumours. Br. J. Cancer 2015, 112, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Deberardinis, R.J. Q&A: Targeting metabolism to diagnose and treat cancer. Cancer Metab. 2014, 2, 5. [Google Scholar] [PubMed]
- Miele, E.; Spinelli, G.P.; Tomao, F.; Zullo, A.; De Marinis, F.; Pasciuti, G.; Rossi, L.; Zoratto, F.; Tomao, S. Positron emission tomography (pet) radiotracers in oncology--utility of 18f-fluoro-deoxy-glucose (fdg)-pet in the management of patients with non-small-cell lung cancer (nsclc). J. Exp. Clin. Cancer Res. 2008, 27, 52. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.N.; Drury, D.R.; Nakada, H.I.; Wolfe, J.B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 1957, 224, 963–969. [Google Scholar] [PubMed]
- Park, G.C.; Kim, J.S.; Roh, J.L.; Choi, S.H.; Nam, S.Y.; Kim, S.Y. Prognostic value of metabolic tumor volume measured by 18f-fdg pet/ct in advanced-stage squamous cell carcinoma of the larynx and hypopharynx. Ann. Oncol 2013, 24, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Sandulache, V.C.; Ow, T.J.; Pickering, C.R.; Frederick, M.J.; Zhou, G.; Fokt, I.; Davis-Malesevich, M.; Priebe, W.; Myers, J.N. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer 2011, 117, 2926–2938. [Google Scholar] [CrossRef] [PubMed]
- Simons, A.L.; Ahmad, I.M.; Mattson, D.M.; Dornfeld, K.J.; Spitz, D.R. 2-deoxy-d-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007, 67, 3364–3370. [Google Scholar] [CrossRef]
- Simons, A.L.; Fath, M.A.; Mattson, D.M.; Smith, B.J.; Walsh, S.A.; Graham, M.M.; Hichwa, R.D.; Buatti, J.M.; Dornfeld, K.; Spitz, D.R. Enhanced response of human head and neck cancer xenograft tumors to cisplatin combined with 2-deoxy-d-glucose correlates with increased 18f-fdg uptake as determined by pet imaging. Int. J. Radiat Oncol. Biol. Phys. 2007, 69, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Celentano, A.; McCullough, M.; Cirillo, N. Glucocorticoids reduce chemotherapeutic effectiveness on oscc cells via glucose-dependent mechanisms. J. Cell Physiol. 2019, 234, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Reckenbeil, J.; Veit, N.; Kuerpig, S.; Meisenheimer, M.; Beier, I.; Stark, H.; Winter, J.; Probstmeier, R. Targeting glucose transport and the nad pathway in tumor cells with stf-31: A re-evaluation. Cell Oncol. 2018, 41, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Jan, C.I.; Tsai, M.H.; Chiu, C.F.; Huang, Y.P.; Liu, C.J.; Chang, N.W. Fenofibrate suppresses oral tumorigenesis via reprogramming metabolic processes: Potential drug repurposing for oral cancer. Int. J. Biol. Sci. 2016, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Yang, J.G.; Liu, Z.J.; Wang, W.M.; Yu, Z.L.; Ren, J.G.; Chen, G.; Zhang, W.; Jia, J. Blockage of glycolysis by targeting pfkfb3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 7. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Li, X.; Luo, Z.; Liu, J.; Fan, Z. Cetuximab reverses the warburg effect by inhibiting hif-1-regulated ldh-a. Mol. Cancer 2013, 12, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of ldh-a by oxamate induces g2/m arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Rodriguez-Cruz, V.; Morris, M.E. Cellular uptake of mct1 inhibitors ar-c155858 and azd3965 and their effects on mct-mediated transport of l-lactate in murine 4t1 breast tumor cancer cells. AAPS J. 2019, 21, 13. [Google Scholar] [CrossRef]
- Mehibel, M.; Ortiz-Martinez, F.; Voelxen, N.; Boyers, A.; Chadwick, A.; Telfer, B.A.; Mueller-Klieser, W.; West, C.M.; Critchlow, S.E.; Williams, K.J.; et al. Statin-induced metabolic reprogramming in head and neck cancer: A biomarker for targeting monocarboxylate transporters. Sci. Rep. 2018, 8, 16804. [Google Scholar] [CrossRef]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.Y.; Wong, L.J. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef] [PubMed]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in human cancer. Int. J. Biochem. Cell Biol. 2009, 41, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The warburg effect: 80 years on. Biochem Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz, D.; Teleman, A.A. Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Rep. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ros in cancer: Initiators, amplifiers or an achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Baek, S.; Shin, D.; Lee, J.; Roh, J.L. Hederagenin induces apoptosis in cisplatin-resistant head and neck cancer cells by inhibiting the nrf2-are antioxidant pathway. Oxid Med. Cell Longev. 2017, 2017, 5498908. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Wu, C.Y.; Shu, C.W.; Wang, S.C.; Chang, M.Y.; Chang, H.W. A novel sulfonyl chromen-4-ones (chw09) preferentially kills oral cancer cells showing apoptosis, oxidative stress, and DNA damage. Env. Toxicol. 2018, 33, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chen, Y.K.; Hsu, Y.L.; Chiu, W.C.; Tsai, C.H.; Hu, S.C.; Hsieh, P.W.; Yuan, S.F. Synthetic beta-nitrostyrene derivative cyt-rx20 as inhibitor of oral cancer cell proliferation and tumor growth through glutathione suppression and reactive oxygen species induction. Head Neck 2017, 39, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.U.; Cho, H.K.; Min, K.J.; Woo, S.M.; Kim, S.; Park, J.W.; Kim, S.H.; Choi, Y.H.; Keum, Y.S.; Hyun, J.W.; et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-flip and mcl-1 expression. Cell Death Dis 2017, 8, e2599. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.Y.; Chueh, F.S.; Ma, Y.S.; Wu, R.S.; Liao, C.L.; Chu, Y.L.; Fan, M.J.; Huang, W.W.; Chung, J.G. Bufalin induced apoptosis in scc4 human tongue cancer cells by decreasing bcl2 and increasing bax expression via the mitochondriadependent pathway. Mol. Med. Rep. 2017, 16, 7959–7966. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.L.; Hung, F.M.; Lee, C.H.; Yeh, M.Y.; Lee, M.H.; Lu, H.F.; Chen, Y.L.; Liu, J.Y.; Chung, J.G. Fisetin induces apoptosis of hsc3 human oral cancer cells through endoplasmic reticulum stress and dysfunction of mitochondria-mediated signaling pathways. In Vivo 2017, 31, 1103–1114. [Google Scholar] [PubMed]
- Chou, G.L.; Peng, S.F.; Liao, C.L.; Ho, H.C.; Lu, K.W.; Lien, J.C.; Fan, M.J.; La, K.C.; Chung, J.G. Casticin impairs cell growth and induces cell apoptosis via cell cycle arrest in human oral cancer scc-4 cells. Env. Toxicol. 2018, 33, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.G.; Jeong, Y.S.; Kim, S.A.; Ahn, S.G. New metformin derivative hl156a prevents oral cancer progression by inhibiting the insulin-like growth factor/akt/mammalian target of rapamycin pathways. Cancer Sci. 2018, 109, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, C.; Yamamoto, K.; Fujiwara-Tani, R.; Luo, Y.; Matsushima, S.; Fujii, K.; Ohmori, H.; Sasahira, T.; Sasaki, T.; Kitadai, Y.; et al. Expression of cytosolic malic enzyme (me1) is associated with disease progression in human oral squamous cell carcinoma. Cancer Sci. 2018, 109, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.H.; Yang, L.P.; Chuang, H.C.; Fitzgerald, A.; Lee, H.Y.; Pickering, C.; Myers, J.N.; Skinner, H.D. Down-regulation of malic enzyme 1 and 2: Sensitizing head and neck squamous cell carcinoma cells to therapy-induced senescence. Head Neck 2016, 38, 934–940. [Google Scholar] [CrossRef]
- Lee, J.; Huang, M.S.; Yang, I.C.; Lai, T.C.; Wang, J.L.; Pang, V.F.; Hsiao, M.; Kuo, M.Y. Essential roles of caspases and their upstream regulators in rotenone-induced apoptosis. Biochem. Biophys. Res. Commun. 2008, 371, 33–38. [Google Scholar] [CrossRef]
- Sandulache, V.C.; Skinner, H.D.; Ow, T.J.; Zhang, A.; Xia, X.; Luchak, J.M.; Wong, L.J.; Pickering, C.R.; Zhou, G.; Myers, J.N. Individualizing antimetabolic treatment strategies for head and neck squamous cell carcinoma based on tp53 mutational status. Cancer 2012, 118, 711–721. [Google Scholar] [CrossRef]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Gao, Z.; Rajthala, S.; Sapkota, D.; Dongre, H.; Parajuli, H.; Suliman, S.; Das, R.; Li, L.; Bindoff, L.A.; et al. Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell Mol. Life Sci. 2019, 1–19. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Gong, Y.; Gu, J.; Lee, J.J.; Lippman, S.M.; Wu, X. Increased leukocyte mitochondrial DNA copy number is associated with oral premalignant lesions: An epidemiology study. Carcinogenesis 2014, 35, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.; Hasegawa, T.; Ueha, T.; Sakakibara, A.; Kawamoto, T.; Minamikawa, T.; Sakai, Y.; Komori, T. Decreased mitochondrial copy numbers in oral squamous cell carcinoma. Head Neck 2016, 38, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. Elife 2016, 5, e10769. [Google Scholar] [CrossRef]
- Kim, M.M.; Clinger, J.D.; Masayesva, B.G.; Ha, P.K.; Zahurak, M.L.; Westra, W.H.; Califano, J.A. Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin. Cancer Res. 2004, 10, 8512–8515. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid metabolic reprogramming in hepatocellular carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S. Lipid metabolism in cancer cachexia. Ann. Palliat. Med. 2019, 8, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Yu, X.H.; Ren, X.H.; Liang, X.H.; Tang, Y.L. Roles of fatty acid metabolism in tumourigenesis: Beyond providing nutrition (review). Mol. Med. Rep. 2018, 18, 5307–5316. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the lipid maps comprehensive classification system for lipids. J. Lipid Res. 2009, 50, 9–14. [Google Scholar] [CrossRef]
- Huang, C.; Freter, C. Lipid metabolism, apoptosis and cancer therapy. Int. J. Mol. Sci. 2015, 16, 924–949. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. Iv. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Schlichtholz, B.; Turyn, J.; Goyke, E.; Biernacki, M.; Jaskiewicz, K.; Sledzinski, Z.; Swierczynski, J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas 2005, 30, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Lupu, R.; Menendez, J.A. Overexpression of fatty acid synthase gene activates her1/her2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008, 41, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic pi3k and k-ras stimulate de novo lipid synthesis through mtorc1 and srebp. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The acetyl-coa carboxylase enzyme: A target for cancer therapy? Expert Rev. Anticancer 2015, 15, 667–676. [Google Scholar] [CrossRef]
- Igal, R.A. Stearoyl-coa desaturase-1: A novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010, 31, 1509–1515. [Google Scholar] [CrossRef]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. Srebp-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef]
- Duan, J.; Sun, L.; Huang, H.; Wu, Z.; Wang, L.; Liao, W. Overexpression of fatty acid synthase predicts a poor prognosis for human gastric cancer. Mol. Med. Rep. 2016, 13, 3027–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.L.; Li, W.C.; Tsai, T.H.; Chiang, H.Y.; Ting, C.T. Body mass index and cholesterol level predict surgical outcome in patients with hepatocellular carcinoma in taiwan - a cohort study. Oncotarget 2016, 7, 22948–22959. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.J.; Siu, L.L.; Chen, E.; Dimitroulakos, J.; Kamel-Reid, S.; Moore, M.J.; Chin, S.; Irish, J.; LaFramboise, S.; Oza, A.M. A phase i trial of prolonged administration of lovastatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck or of the cervix. Eur. J. Cancer 2005, 41, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Takeda, I.; Maruya, S.; Shirasaki, T.; Mizukami, H.; Takahata, T.; Myers, J.N.; Kakehata, S.; Yagihashi, S.; Shinkawa, H. Simvastatin inactivates beta1-integrin and extracellular signal-related kinase signaling and inhibits cell proliferation in head and neck squamous cell carcinoma cells. Cancer Sci. 2007, 98, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, A.; Kurnaz, S.C. An investigation of oxidative stress and coenzyme q10 levels in patients with head and neck squamous cell carcinomas. Eur. Arch. Otorhinolaryngol. 2019, 276, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bhatt, M.L.; Misra, M.K. Lipid peroxidation and antioxidant status in head and neck squamous cell carcinoma patients. Oxid Med. Cell Longev. 2009, 2, 68–72. [Google Scholar] [CrossRef]
- Rasheed, M.H.; Beevi, S.S.; Geetha, A. Enhanced lipid peroxidation and nitric oxide products with deranged antioxidant status in patients with head and neck squamous cell carcinoma. Oral Oncol. 2007, 43, 333–338. [Google Scholar] [CrossRef]
- Chan, L.P.; Chou, T.H.; Ding, H.Y.; Chen, P.R.; Chiang, F.Y.; Kuo, P.L.; Liang, C.H. Apigenin induces apoptosis via tumor necrosis factor receptor- and bcl-2-mediated pathway and enhances susceptibility of head and neck squamous cell carcinoma to 5-fluorouracil and cisplatin. Biochim. Biophys Acta. 2012, 1820, 1081–1091. [Google Scholar] [CrossRef]
- Huang, Y.; Bell, L.N.; Okamura, J.; Kim, M.S.; Mohney, R.P.; Guerrero-Preston, R.; Ratovitski, E.A. Phospho-deltanp63alpha/srebf1 protein interactions: Bridging cell metabolism and cisplatin chemoresistance. Cell Cycle 2012, 11, 3810–3827. [Google Scholar] [CrossRef]
- Su, Y.W.; Lin, Y.H.; Pai, M.H.; Lo, A.C.; Lee, Y.C.; Fang, I.C.; Lin, J.; Hsieh, R.K.; Chang, Y.F.; Chen, C.L. Association between phosphorylated amp-activated protein kinase and acetyl-coa carboxylase expression and outcome in patients with squamous cell carcinoma of the head and neck. PLoS ONE 2014, 9, e96183. [Google Scholar] [CrossRef]
- Luo, J.; Hong, Y.; Lu, Y.; Qiu, S.; Chaganty, B.K.; Zhang, L.; Wang, X.; Li, Q.; Fan, Z. Acetyl-coa carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the warburg effect by cetuximab. Cancer Lett. 2017, 384, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, C.; Chen, L.; Wang, P.; Fang, Y.; Zhu, J.; Chen, S.; Du, J.; Shen, B.; Wu, K.; et al. The role of acetyl-coa carboxylase2 in head and neck squamous cell carcinoma. Peer J. 2019, 7, e7037. [Google Scholar] [CrossRef] [PubMed]
- Mims, J.; Bansal, N.; Bharadwaj, M.S.; Chen, X.; Molina, A.J.; Tsang, A.W.; Furdui, C.M. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat Res. 2015, 183, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, S.; Agarwal, R.; Goel, S.K.; Panday, R.K.; Singh, R.; Sukumaran, R.; Khare, S.; Kumar, A. Altered expression of sphingosine-1-phosphate metabolizing enzymes in oral cancer correlate with clinicopathological attributes. Cancer Invest. 2017, 35, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Kakuki, T.; Obata, K.; Nomura, K.; Miyata, R.; Kondo, A.; Kurose, M.; Kakiuchi, A.; Kaneko, Y.; Kohno, T.; et al. The behavior and role of lipolysis-stimulated lipoprotein receptor, a component of tricellular tight junctions, in head and neck squamous cell carcinomas. Anticancer Res. 2016, 36, 5895–5904. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Peng, J.; Chen, X.; Li, H.; Song, M.; Cheng, B.; Wu, T. Obesity and genes related to lipid metabolism predict poor survival in oral squamous cell carcinoma. Oral Oncol 2019, 89, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Coloff, J.L. The diverse functions of non-essential amino acids in cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef]
- Ide, A.L.; Portari, G.V.; Padovan, G.J.; Rosa, F.T.; Mello-Filho, F.V.; Marchini, J.S. Amino acids in squamous cell carcinomas and adjacent normal tissues from patients with larynx and oral cavity lesions. Clin. (Sao Paulo) 2012, 67, 1225–1227. [Google Scholar]
- Yonezawa, K.; Nishiumi, S.; Kitamoto-Matsuda, J.; Fujita, T.; Morimoto, K.; Yamashita, D.; Saito, M.; Otsuki, N.; Irino, Y.; Shinohara, M.; et al. Serum and tissue metabolomics of head and neck cancer. Cancer Genom. Proteom. 2013, 10, 233–238. [Google Scholar]
- Bothwell, P.J.; Kron, C.D.; Wittke, E.F.; Czerniak, B.N.; Bode, B.P. Targeted suppression and knockout of asct2 or lat1 in epithelial and mesenchymal human liver cancer cells fail to inhibit growth. Int. J. Mol. Sci. 2018, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Hafliger, P.; Charles, R.P. The l-type amino acid transporter lat1-an emerging target in cancer. Int. J. Mol. Sci. 2019, 20, 2093. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Oriuchi, N.; Imai, H.; Shimizu, K.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Tanaka, S.; Ishizuka, T.; Kanai, Y.; et al. Prognostic significance of l-type amino acid transporter 1 expression in resectable stage i-iii nonsmall cell lung cancer. Br. J. Cancer 2008, 98, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Sunose, Y.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; Itoh, H.; Nagamori, S.; et al. Prognostic significance of l-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Br. J. Cancer 2012, 107, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Maeda, K.; Ohtani, H.; Nagahara, H.; Shibutani, M.; Hirakawa, K. Expression of xct as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015, 35, 677–682. [Google Scholar] [PubMed]
- Wang, J.; Papanicolau-Sengos, A.; Chintala, S.; Wei, L.; Liu, B.; Hu, Q.; Miles, K.M.; Conroy, J.M.; Glenn, S.T.; Costantini, M.; et al. Collecting duct carcinoma of the kidney is associated with cdkn2a deletion and slc family gene up-regulation. Oncotarget 2016, 7, 29901–29915. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, X.; Song, B.; Qiu, X.; Zhao, J. Mir-375/slc7a11 axis regulates oral squamous cell carcinoma proliferation and invasion. Cancer Med. 2017, 6, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, A.; Kim, M.; Kaira, K.; Miyashita, G.; Negishi, A.; Oriuchi, N.; Higuchi, T.; Tsushima, Y.; Kanai, Y.; Yokoo, S.; et al. Diagnostic usefulness of (1)(8)f-famt pet and l-type amino acid transporter 1 (lat1) expression in oral squamous cell carcinoma. Eur. J. Nucl Med. Mol. Imaging 2013, 40, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Kimura, T.; Yamaga, T.; Kawada, A.; Ochiai, T.; Endou, H.; Sakurai, H. Metformin enhances anti-tumor effect of l-type amino acid transporter 1 (lat1) inhibitor. J. Pharm. Sci. 2016, 131, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Moriya, S.; Kazama, H.; Hirasawa, K.; Miyahara, K.; Kokuba, H.; Hino, H.; Kikuchi, H.; Takano, N.; Hiramoto, M.; et al. Amino acid starvation culture condition sensitizes egfr-expressing cancer cell lines to gefitinib-mediated cytotoxicity by inducing atypical necroptosis. Int. J. Oncol. 2018, 52, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Lu, Y.; Qiu, S.; Wang, Y.; Qin, J.; Fan, Z. Ap1g1 is involved in cetuximab-mediated downregulation of asct2-egfr complex and sensitization of human head and neck squamous cell carcinoma cells to ros-induced apoptosis. Cancer Lett. 2017, 408, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. Xct inhibition depletes cd44v-expressing tumor cells that are resistant to egfr-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, W.; Wei, Z.; Xu, L.I.; Yang, X.; Du, Y. Xct expression modulates cisplatin resistance in tca8113 tongue carcinoma cells. Oncol Lett. 2016, 12, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hamstra, D.A.; Lee, K.C.; Eisbruch, A.; Sunkara, P.; Borgonha, S.; Phillip, B.; Campbell, K.C.M.; Ross, B.D.; Rehemtulla, A. Double-blind placebo-controlled multicenter phase ii trial to evaluate d-methionine in preventing/reducing oral mucositis induced by radiation and chemotherapy for head and neck cancer. Head Neck 2018, 40, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Vsiansky, V.; Svobodova, M.; Gumulec, J.; Cernei, N.; Sterbova, D.; Zitka, O.; Kostrica, R.; Smilek, P.; Plzak, J.; Betka, J.; et al. Prognostic significance of serum free amino acids in head and neck cancers. Cells 2019, 8, 428. [Google Scholar] [CrossRef]
- Huang, C.C.; Tsai, S.T.; Kuo, C.C.; Chang, J.S.; Jin, Y.T.; Chang, J.Y.; Hsiao, J.R. Arginine deprivation as a new treatment strategy for head and neck cancer. Oral Oncol. 2012, 48, 1227–1235. [Google Scholar] [CrossRef]
- Tomlinson, B.K.; Thomson, J.A.; Bomalaski, J.S.; Diaz, M.; Akande, T.; Mahaffey, N.; Li, T.; Dutia, M.P.; Kelly, K.; Gong, I.Y.; et al. Phase i trial of arginine deprivation therapy with adi-peg 20 plus docetaxel in patients with advanced malignant solid tumors. Clin. Cancer Res. 2015, 21, 2480–2486. [Google Scholar] [CrossRef]
- Kamarajan, P.; Rajendiran, T.M.; Kinchen, J.; Bermudez, M.; Danciu, T.; Kapila, Y.L. Head and neck squamous cell carcinoma metabolism draws on glutaminolysis, and stemness is specifically regulated by glutaminolysis via aldehyde dehydrogenase. J. Proteome Res. 2017, 16, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Guo, Y.; Seo, W.; Zhang, R.; Lu, C.; Wang, Y.; Luo, L.; Paul, B.; Yan, W.; Saxena, D.; et al. Targeting cellular metabolism to reduce head and neck cancer growth. Sci. Rep. 2019, 9, 4995. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [PubMed]
- Esteller, M. Non-coding rnas in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Denaro, N.; Merlano, M.C.; Russi, E.G.; Lo Nigro, C. Non coding rnas in head and neck squamous cell carcinoma (hnscc): A clinical perspective. Anticancer Res. 2014, 34, 6887–6896. [Google Scholar] [PubMed]
- Guglas, K.; Bogaczynska, M.; Kolenda, T.; Rys, M.; Teresiak, A.; Blizniak, R.; Lasinska, I.; Mackiewicz, J.; Lamperska, K. Lncrna in hnscc: Challenges and potential. Contemp Oncol. (Pozn) 2017, 21, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Wei, Y.Y.; Yang, C.C.; Liu, C.J.; Yeh, L.Y.; Chou, C.H.; Chang, K.W.; Lin, S.C. Mir-125b suppresses oral oncogenicity by targeting the anti-oxidative gene prxl2a. Redox Biol. 2019, 22, 101140. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Yang, C.C.; Kao, S.Y.; Liu, C.J.; Lin, S.C.; Chang, K.W. Microrna-211 enhances the oncogenicity of carcinogen-induced oral carcinoma by repressing tcf12 and increasing antioxidant activity. Cancer Res. 2016, 76, 4872–4886. [Google Scholar] [CrossRef] [PubMed]
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding rna uca1 promotes glycolysis by upregulating hexokinase 2 through the mtor-stat3/microrna143 pathway. Cancer Sci. 2014, 105, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long noncoding rna hulc modulates abnormal lipid metabolism in hepatoma cells through an mir-9-mediated rxra signaling pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zhang, Y.; Chen, Y.; Bai, Y.; Zhang, Y. Long noncoding rna linc01234 promotes serine hydroxymethyltransferase 2 expression and proliferation by competitively binding mir-642a-5p in colon cancer. Cell Death Dis. 2019, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. Lncrna-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ren, J.; Zhang, D.; Li, Y.; Huang, X.; Hu, Q.; Wang, H.; Song, Y.; Ni, Y.; Hou, Y. A novel stromal lncrna signature reprograms fibroblasts to promote the growth of oral squamous cell carcinoma via lncrna-caf/interleukin-33. Carcinogenesis 2018, 39, 397–406. [Google Scholar] [CrossRef]
- Xu, S.; Guo, J.; Zhang, W. Lncrna pcat19 promotes the proliferation of laryngocarcinoma cells via modulation of the mir-182/pdk4 axis. J. Cell Biochem. 2019, 120, 12810–12821. [Google Scholar] [CrossRef]
- Shih, J.W.; Chiang, W.F.; Wu, A.T.H.; Wu, M.H.; Wang, L.Y.; Yu, Y.L.; Hung, Y.W.; Wang, W.C.; Chu, C.Y.; Hung, C.L.; et al. Long noncoding rna lnchifcar/mir31hg is a hif-1alpha co-activator driving oral cancer progression. Nat. Commun. 2017, 8, 15874. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, S.; Chen, J.; Wang, Z.; Liang, X.; Wang, X.; Jiang, J.; Lang, J.; Li, L. Long noncoding rna has2-as1 mediates hypoxia-induced invasiveness of oral squamous cell carcinoma. Mol. Carcinog. 2017, 56, 2210–2222. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Calin, G.A.; Lopez-Berestein, G.; Sood, A.K. Mirna deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 2016, 6, 235–246. [Google Scholar] [CrossRef]
- Kumar, B.; Yadav, A.; Lang, J.; Teknos, T.N.; Kumar, P. Dysregulation of microrna-34a expression in head and neck squamous cell carcinoma promotes tumor growth and tumor angiogenesis. PLoS ONE 2012, 7, e37601. [Google Scholar] [CrossRef]
- Li, Y.Y.; Tao, Y.W.; Gao, S.; Li, P.; Zheng, J.M.; Zhang, S.E.; Liang, J.; Zhang, Y. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine mir-34a-5p. EBioMedicine 2018, 36, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor mir-34a targets pd-l1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Rapado-Gonzalez, O.; Majem, B.; Muinelo-Romay, L.; Alvarez-Castro, A.; Santamaria, A.; Gil-Moreno, A.; Lopez-Lopez, R.; Suarez-Cunqueiro, M.M. Human salivary micrornas in cancer. J. Cancer 2018, 9, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Camps, C.; Buffa, F.M.; Colella, S.; Moore, J.; Sotiriou, C.; Sheldon, H.; Harris, A.L.; Gleadle, J.M.; Ragoussis, J. Hsa-mir-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin. Cancer Res. 2008, 14, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.S.; Huang, X.; Cao, H.; Christman-Skieller, C.; Bennewith, K.; Le, Q.T.; Koong, A.C. Circulating mir-210 as a novel hypoxia marker in pancreatic cancer. Transl. Oncol. 2010, 3, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.T.; Giaccia, A.J. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.L.; et al. Microrna-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein iscu. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef]
- Saenz-de-Santa-Maria, I.; Bernardo-Castineira, C.; Secades, P.; Bernaldo-de-Quiros, S.; Rodrigo, J.P.; Astudillo, A.; Chiara, M.D. Clinically relevant hif-1alpha-dependent metabolic reprogramming in oropharyngeal squamous cell carcinomas includes coordinated activation of caix and the mir-210/iscu signaling axis, but not mct1 and mct4 upregulation. Oncotarget 2017, 8, 13730–13746. [Google Scholar] [CrossRef]
- Xu, P.; Li, Y.; Zhang, H.; Li, M.; Zhu, H. Microrna-340 mediates metabolic shift in oral squamous cell carcinoma by targeting glucose transporter-1. J. Oral Maxillofac Surg. 2016, 74, 844–850. [Google Scholar] [CrossRef]
- Yoshizawa, J.M.; Wong, D.T. Salivary micrornas and oral cancer detection. Methods Mol. Biol. 2013, 936, 313–324. [Google Scholar]
- Schneider, A.; Victoria, B.; Lopez, Y.N.; Suchorska, W.; Barczak, W.; Sobecka, A.; Golusinski, W.; Masternak, M.M.; Golusinski, P. Tissue and serum microrna profile of oral squamous cell carcinoma patients. Sci. Rep. 2018, 8, 675. [Google Scholar] [CrossRef] [PubMed]
- Chandra Gupta, S.; Nandan Tripathi, Y. Potential of long non-coding rnas in cancer patients: From biomarkers to therapeutic targets. Int. J. Cancer 2017, 140, 1955–1967. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Qiang, C.; Gao, L.; Li, S.M.; Zhang, L.M.; Wang, X.L.; Dong, J.W.; Chen, C.; Liu, C.Y.; Zhi, K.Q. Circulating microrna-21 (mir-21) and phosphatase and tensin homolog (pten) are promising novel biomarkers for detection of oral squamous cell carcinoma. Biomarkers 2014, 19, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Hung, P.S.; Liu, C.J.; Chou, C.S.; Kao, S.Y.; Yang, C.C.; Chang, K.W.; Chiu, T.H.; Lin, S.C. Mir-146a enhances the oncogenicity of oral carcinoma by concomitant targeting of the irak1, traf6 and numb genes. PLoS ONE 2013, 8, e79926. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wu, Z.; Zhang, J.; Su, B. Salivary lncrna as a potential marker for oral squamous cell carcinoma diagnosis. Mol. Med. Rep. 2013, 7, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Park, N.J.; Zhou, H.; Elashoff, D.; Henson, B.S.; Kastratovic, D.A.; Abemayor, E.; Wong, D.T. Salivary microrna: Discovery, characterization, and clinical utility for oral cancer detection. Clin. Cancer Res. 2009, 15, 5473–5477. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Ye, P.; Zhang, W.; Rao, J.; Xie, Z. diagnostic values of salivary versus and plasma microrna-21 for early esophageal cancer. J. Southern Med. University 2014, 34, 885–889. [Google Scholar]
- Xie, Z.J.; Chen, G.; Zhang, X.C.; Li, D.F.; Huang, J.; Li, Z.J. Saliva supernatant mir-21: A novel potential biomarker for esophageal cancer detection. Asian Pac. J. Cancer Prev. 2012, 13, 6145–6149. [Google Scholar] [CrossRef]
- Liu, C.J.; Kao, S.Y.; Tu, H.F.; Tsai, M.M.; Chang, K.W.; Lin, S.C. Increase of microrna mir-31 level in plasma could be a potential marker of oral cancer. Oral Dis. 2010, 16, 360–364. [Google Scholar] [CrossRef]
- Liu, C.J.; Lin, S.C.; Yang, C.C.; Cheng, H.W.; Chang, K.W. Exploiting salivary mir-31 as a clinical biomarker of oral squamous cell carcinoma. Head Neck 2012, 34, 219–224. [Google Scholar] [CrossRef]
- Kao, Y.Y.; Tu, H.F.; Kao, S.Y.; Chang, K.W.; Lin, S.C. The increase of oncogenic mirna expression in tongue carcinogenesis of a mouse model. Oral Oncol. 2015, 51, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, X.; Liu, X.; Yu, Y.; Pan, H.; Haak, R.; Schmidt, J.; Ziebolz, D.; Schmalz, G. Complex integrated analysis of lncrnas-mirnas-mrnas in oral squamous cell carcinoma. Oral Oncol. 2017, 73, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.L.; Liu, Y.H.; Ban, J.D.; Wang, W.J.; Han, M.; Kong, P.; Li, B.H. Pathway analysis of a genomewide association study on a long noncoding rna expression profile in oral squamous cell carcinoma. Oncol. Rep. 2019, 41, 895–907. [Google Scholar] [PubMed]
- Curry, J.M.; Sprandio, J.; Cognetti, D.; Luginbuhl, A.; Bar-ad, V.; Pribitkin, E.; Tuluc, M. Tumor microenvironment in head and neck squamous cell carcinoma. Semin. Oncol. 2014, 41, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Freiser, M.E.; Serafini, P.; Weed, D.T. The immune system and head and neck squamous cell carcinoma: From carcinogenesis to new therapeutic opportunities. Immunol. Res. 2013, 57, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Z.; South, A.P. Tumour-stroma crosstalk in the development of squamous cell carcinoma. Int. J. Biochem. Cell Biol. 2014, 53, 450–458. [Google Scholar] [CrossRef]
- Schmitz, S.; Machiels, J.P. Targeting the tumor environment in squamous cell carcinoma of the head and neck. Curr. Treat. Options Oncol. 2016, 17, 37. [Google Scholar] [CrossRef]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: A systematic review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef]
- Eisma, R.J.; Spiro, J.D.; Kreutzer, D.L. Vascular endothelial growth factor expression in head and neck squamous cell carcinoma. Am. J. Surg. 1997, 174, 513–517. [Google Scholar] [CrossRef]
- Mark, R.; Bermejo, J.L.; Bierhaus, A.; Plinkert, P.K.; Angel, P.; Hess, J. The receptor for advanced glycation end products is dispensable in a mouse model of oral and esophageal carcinogenesis. Histol. Histopathol. 2013, 28, 1585–1594. [Google Scholar]
- Kartha, V.K.; Stawski, L.; Han, R.; Haines, P.; Gallagher, G.; Noonan, V.; Kukuruzinska, M.; Monti, S.; Trojanowska, M. Pdgfrbeta is a novel marker of stromal activation in oral squamous cell carcinomas. PLoS ONE 2016, 11, e0154645. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Bhola, N.E.; Grandis, J.R. Hgf/met signaling in head and neck cancer: Impact on the tumor microenvironment. Clin. Cancer Res. 2016, 22, 4005–4013. [Google Scholar] [CrossRef] [PubMed]
- Szturz, P.; Budikova, M.; Vermorken, J.B.; Horova, I.; Gal, B.; Raymond, E.; de Gramont, A.; Faivre, S. Prognostic value of c-met in head and neck cancer: A systematic review and meta-analysis of aggregate data. Oral Oncol. 2017, 74, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Szturz, P.; Raymond, E.; Abitbol, C.; Albert, S.; de Gramont, A.; Faivre, S. Understanding c-met signalling in squamous cell carcinoma of the head & neck. Crit. Rev. Oncol. Hematol. 2017, 111, 39–51. [Google Scholar] [PubMed]
- Pal, S.K.; Nguyen, C.T.; Morita, K.I.; Miki, Y.; Kayamori, K.; Yamaguchi, A.; Sakamoto, K. Thbs1 is induced by tgfb1 in the cancer stroma and promotes invasion of oral squamous cell carcinoma. J. Oral Pathol. Med. 2016, 45, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zhao, Z.L.; Yu, G.T.; Wu, L.; Deng, W.W.; Li, Y.C.; Liu, J.F.; Bu, L.L.; Liu, B.; Kulkarni, A.B.; et al. Gamma-secretase inhibitor reduces immunosuppressive cells and enhances tumour immunity in head and neck squamous cell carcinoma. Int. J. Cancer 2018, 142, 999–1009. [Google Scholar] [CrossRef]
- Le, P.N.; Keysar, S.B.; Miller, B.; Eagles, J.R.; Chimed, T.S.; Reisinger, J.; Gomez, K.E.; Nieto, C.; Jackson, B.C.; Somerset, H.L.; et al. Wnt signaling dynamics in head and neck squamous cell cancer tumor-stroma interactions. Mol. Carcinog. 2019, 58, 398–410. [Google Scholar] [CrossRef]
- Chen, Y.C.; Cheng, Y.H.; Kim, H.S.; Ingram, P.N.; Nor, J.E.; Yoon, E. Paired single cell co-culture microenvironments isolated by two-phase flow with continuous nutrient renewal. Lab. Chip. 2014, 14, 2941–2947. [Google Scholar] [CrossRef] [Green Version]
- Bisetto, S.; Whitaker-Menezes, D.; Wilski, N.A.; Tuluc, M.; Curry, J.; Zhan, T.; Snyder, C.M.; Martinez-Outschoorn, U.E.; Philp, N.J. Monocarboxylate transporter 4 (mct4) knockout mice have attenuated 4nqo induced carcinogenesis; a role for mct4 in driving oral squamous cell cancer. Front. Oncol. 2018, 8, 324. [Google Scholar] [CrossRef]
- Ohashi, T.; Aoki, M.; Tomita, H.; Akazawa, T.; Sato, K.; Kuze, B.; Mizuta, K.; Hara, A.; Nagaoka, H.; Inoue, N.; et al. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci. 2017, 108, 1128–1134. [Google Scholar] [CrossRef]
- Curry, J.; Tassone, P.; Gill, K.; Tuluc, M.; BarAd, V.; Mollaee, M.; Whitaker-Menezes, D.; Rodeck, U.; Luginbuhl, A.; Cognetti, D.; et al. Tumor metabolism in the microenvironment of nodal metastasis in oral squamous cell carcinoma. Otolaryngol. Head Neck Surg. 2017, 157, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Krupar, R.; Robold, K.; Gaag, D.; Spanier, G.; Kreutz, M.; Renner, K.; Hellerbrand, C.; Hofstaedter, F.; Bosserhoff, A.K. Immunologic and metabolic characteristics of hpv-negative and hpv-positive head and neck squamous cell carcinomas are strikingly different. Virchows Arch. 2014, 465, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Na, K.J.; Choi, H. Tumor metabolic features identified by (18)f-fdg pet correlate with gene networks of immune cell microenvironment in head and neck cancer. J. Nucl. Med. 2018, 59, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, E.; McCrory, A.; Talbert, M.; Young, G.; Murphy-Ullrich, J.; Gladson, C. Elevated expression of tgf-beta1 in head and neck cancer-associated fibroblasts. Mol. Carcinog. 2004, 40, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Metzler, V.M.; Pritz, C.; Riml, A.; Romani, A.; Tuertscher, R.; Steinbichler, T.; Dejaco, D.; Riechelmann, H.; Dudas, J. Separation of cell survival, growth, migration, and mesenchymal transdifferentiation effects of fibroblast secretome on tumor cells of head and neck squamous cell carcinoma. Tumour Biol. 2017, 39, 1010428317705507. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Shi, H.; Lin, F.; Dasari, S.; Bednash, J.; Thorne, S.; Watkins, S.; Joshi, R.; Thomas, S.M. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head Neck 2014, 36, 385–392. [Google Scholar] [CrossRef]
- Rasanen, K.; Sriswasdi, S.; Valiga, A.; Tang, H.Y.; Zhang, G.; Perego, M.; Somasundaram, R.; Li, L.; Speicher, K.; Klein-Szanto, A.J.; et al. Comparative secretome analysis of epithelial and mesenchymal subpopulations of head and neck squamous cell carcinoma identifies s100a4 as a potential therapeutic target. Mol. Cell Proteom. 2013, 12, 3778–3792. [Google Scholar] [CrossRef]
- Jiang, E.; Xu, Z.; Wang, M.; Yan, T.; Huang, C.; Zhou, X.; Liu, Q.; Wang, L.; Chen, Y.; Wang, H.; et al. Tumoral microvesicle-activated glycometabolic reprogramming in fibroblasts promotes the progression of oral squamous cell carcinoma. FASEB J. 2019, 33, 5690–5703. [Google Scholar] [CrossRef]
- Reichert, T.E.; Strauss, L.; Wagner, E.M.; Gooding, W.; Whiteside, T.L. Signaling abnormalities, apoptosis, and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin. Cancer Res. 2002, 8, 3137–3145. [Google Scholar]
- Strauss, L.; Bergmann, C.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. The frequency and suppressor function of cd4+cd25highfoxp3+ t cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2007, 13, 6301–6311. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-gamma drives treg fragility to promote anti-tumor immunity. Cell 2017, 169, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Kesselring, R.; Thiel, A.; Pries, R.; Trenkle, T.; Wollenberg, B. Human th17 cells can be induced through head and neck cancer and have a functional impact on hnscc development. Br. J. Cancer 2010, 103, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Nieh, S.; Jao, S.W.; Wu, M.Z.; Liu, C.L.; Chang, Y.C.; Lin, Y.S. The paracrine effect of cancer-associated fibroblast-induced interleukin-33 regulates the invasiveness of head and neck squamous cell carcinoma. J. Pathol. 2013, 231, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Yaguchi, T.; Ohmura, G.; Ohta, S.; Kobayashi, A.; Kawamura, N.; Fujita, T.; Nakano, H.; Shimada, T.; Takahashi, T.; et al. Autocrine and paracrine loops between cancer cells and macrophages promote lymph node metastasis via ccr4/ccl22 in head and neck squamous cell carcinoma. Int. J. Cancer 2013, 132, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.J.; Westerkamp, A.M.; Kansy, B.A.; Bruderek, K.; Dissmann, P.A.; Dumitru, C.A.; Lang, S.; Jackson, E.K.; Brandau, S. Adenosine metabolism of human mesenchymal stromal cells isolated from patients with head and neck squamous cell carcinoma. Immunobiology 2017, 222, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jie, H.B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. Pd-1/shp-2 inhibits tc1/th1 phenotypic responses and the activation of t cells in the tumor microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef]
- Wen, Y.H.; Lin, H.Q.; Li, H.; Zhao, Y.; Lui, V.W.Y.; Chen, L.; Wu, X.M.; Sun, W.; Wen, W.P. Stromal interleukin-33 promotes regulatory t cell-mediated immunosuppression in head and neck squamous cell carcinoma and correlates with poor prognosis. Cancer Immunol. Immunother 2019, 68, 221–232. [Google Scholar] [CrossRef]
- Nguyen, N.; Bellile, E.; Thomas, D.; McHugh, J.; Rozek, L.; Virani, S.; Peterson, L.; Carey, T.E.; Walline, H.; Moyer, J.; et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck 2016, 38, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Gross, N.; Liu, Y.; Zhang, L.; Li, G.; Huang, Z.; Yang, J. A high ratio of il-12rbeta2-positive tumor-infiltrating lymphocytes indicates favorable prognosis in laryngeal cancer. Oral Oncol 2017, 74, 148–156. [Google Scholar] [CrossRef]
- Wallis, S.P.; Stafford, N.D.; Greenman, J. Clinical relevance of immune parameters in the tumor microenvironment of head and neck cancers. Head Neck 2015, 37, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, K.; Takahashi, H.; Kaira, K.; Toyoda, M.; Murata, T.; Ohnishi, H.; Oyama, T.; Chikamatsu, K. Relationship between tumor-associated macrophage subsets and cd47 expression in squamous cell carcinoma of the head and neck in the tumor microenvironment. Lab. Invest. 2016, 96, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Troiano, G.; Caponio, V.C.A.; Zhurakivska, K.; Arena, C.; Pannone, G.; Mascitti, M.; Santarelli, A.; Lo Muzio, L. High pd-l1 expression in the tumour cells did not correlate with poor prognosis of patients suffering for oral squamous cells carcinoma: A meta-analysis of the literature. Cell Prolif 2019, 52, e12537. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Lartigau, E.; Le Ridant, A.M.; Lambin, P.; Weeger, P.; Martin, L.; Sigal, R.; Lusinchi, A.; Luboinski, B.; Eschwege, F.; Guichard, M. Oxygenation of head and neck tumors. Cancer 1993, 71, 2319–2325. [Google Scholar] [CrossRef]
- Zhu, G.; Tang, Y.; Geng, N.; Zheng, M.; Jiang, J.; Li, L.; Li, K.; Lei, Z.; Chen, W.; Fan, Y.; et al. Hif-alpha/mif and nf-kappab/il-6 axes contribute to the recruitment of cd11b+gr-1+ myeloid cells in hypoxic microenvironment of hnscc. Neoplasia 2014, 16, 168–179. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Wang, S.; Wang, Z.; Jiang, J.; Wang, W.; Li, X.; Chen, J.; Liu, K.; Li, C.; et al. Exosomes derived from hypoxic oral squamous cell carcinoma cells deliver mir-21 to normoxic cells to elicit a prometastatic phenotype. Cancer Res. 2016, 76, 1770–1780. [Google Scholar] [CrossRef]
- Mayer, A.; Zahnreich, S.; Brieger, J.; Vaupel, P.; Schmidberger, H. Downregulation of egfr in hypoxic, diffusion-limited areas of squamous cell carcinomas of the head and neck. Br. J. Cancer 2016, 115, 1351–1358. [Google Scholar] [CrossRef]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A role for p38 mapk in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol. Oncol. 2014, 8, 105–118. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | ncRNA | Regulating Target/Role | Molecular Alterations | Reference |
|---|---|---|---|---|
| Bladder cancer | LncUCA1 | glucose metabolism | hexokinase 2 (HK2)↑, mTOR-STAT3↑ | [151] |
| Hepatocellular carcinoma | LncHULC | lipid metabolism | ACSL1↑ | [152] |
| Colon cancer | LncRNA LINC01234 | amino acid metabolism | miR-642a↓ | [153] |
| Head and neck cancer | LncCAF (FLJ22447) | oncogenic | IL-33↑ | [155] |
| Head and neck cancer | LncZFAS1 | tumor suppressive | EGFR↓, PD-L1↓ | [156] |
| Head and neck cancer | LncHIFCAR (MIR31HG) | glucose metabolism | HIF-1↑ | [157] |
| Head and neck cancer | ncHAS2-AS1 | hypoxia | [158] | |
| Larynx carcinoma | LncPCAT19 | mitochondria | miR-182↑ | [156] |
| Head and neck cancer | miR-34a | tumor suppressive | VEGF↓, AXL↓ | [160,161] |
| Acute myeloid leukemia | miR-34a | PD-L1↓ | [162] | |
| Pancreatic ductal adenocarcinoma | miR-34a | [163] | ||
| Oropharyngeal squamous cell carcinoma | miR-210 | ISCU↓ | [168] | |
| Head and neck cancer | miR-340 | glucose metabolism | GLUT1↑ | [169] |
| Head and neck cancer | miR-21, LncRNAs HOXA11-AS, LINC00964 and MALAT-1 | plasma biomarker | [173] | |
| Head and neck cancer | miR-146a | plasma biomarker | [174] | |
| Head and neck cancer | MALAT-1, HOTAIR, NEAT-1, HULC, MEG-3 and UCA1 | oncogenic | [175] | |
| Head and neck cancer | miR-125a and miR-200a | salivary marker | [176] | |
| Esophageal cancer | miR-21 | salivary marker | [177,178] | |
| Head and neck cancer | miR-31 | plasma and salivary biomarker | [179,180] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, Y.-T.; Chen, Y.-F.; Lin, S.-C.; Chang, K.-W.; Li, W.-C. Targeting Cellular Metabolism Modulates Head and Neck Oncogenesis. Int. J. Mol. Sci. 2019, 20, 3960. https://doi.org/10.3390/ijms20163960
Hsieh Y-T, Chen Y-F, Lin S-C, Chang K-W, Li W-C. Targeting Cellular Metabolism Modulates Head and Neck Oncogenesis. International Journal of Molecular Sciences. 2019; 20(16):3960. https://doi.org/10.3390/ijms20163960
Chicago/Turabian StyleHsieh, Yi-Ta, Yi-Fen Chen, Shu-Chun Lin, Kuo-Wei Chang, and Wan-Chun Li. 2019. "Targeting Cellular Metabolism Modulates Head and Neck Oncogenesis" International Journal of Molecular Sciences 20, no. 16: 3960. https://doi.org/10.3390/ijms20163960
APA StyleHsieh, Y. -T., Chen, Y. -F., Lin, S. -C., Chang, K. -W., & Li, W. -C. (2019). Targeting Cellular Metabolism Modulates Head and Neck Oncogenesis. International Journal of Molecular Sciences, 20(16), 3960. https://doi.org/10.3390/ijms20163960