Pan-HDAC Inhibitors Promote Tau Aggregation by Increasing the Level of Acetylated Tau


 ,
, 
Abstract
:1. Introduction
2. Results
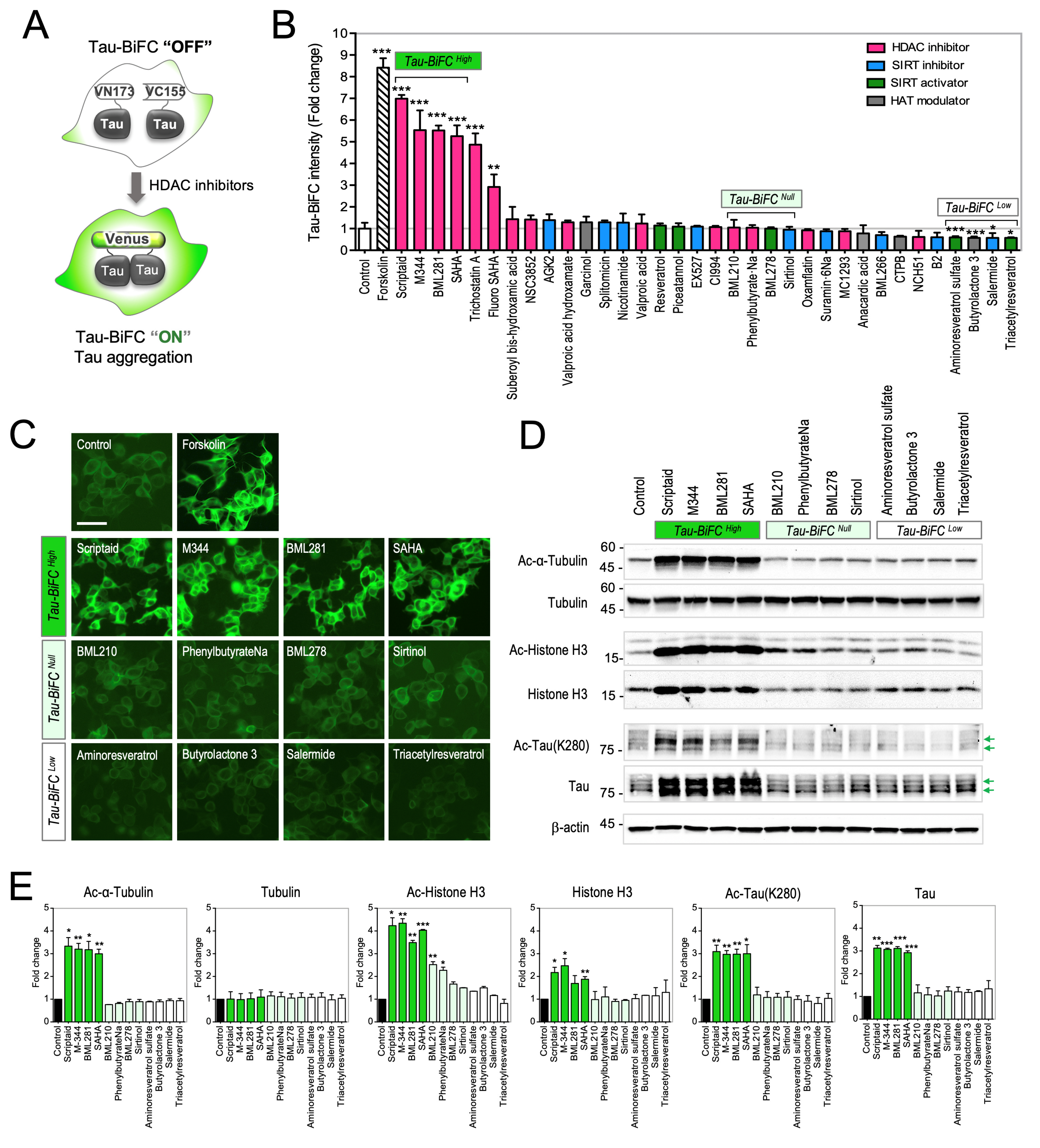
2.1. Evaluation of HDAC Modulators on Tau-BiFC Sensor
2.2. Activation of Tau Pathology by the Treatment of Pan-HDAC Inhibitors
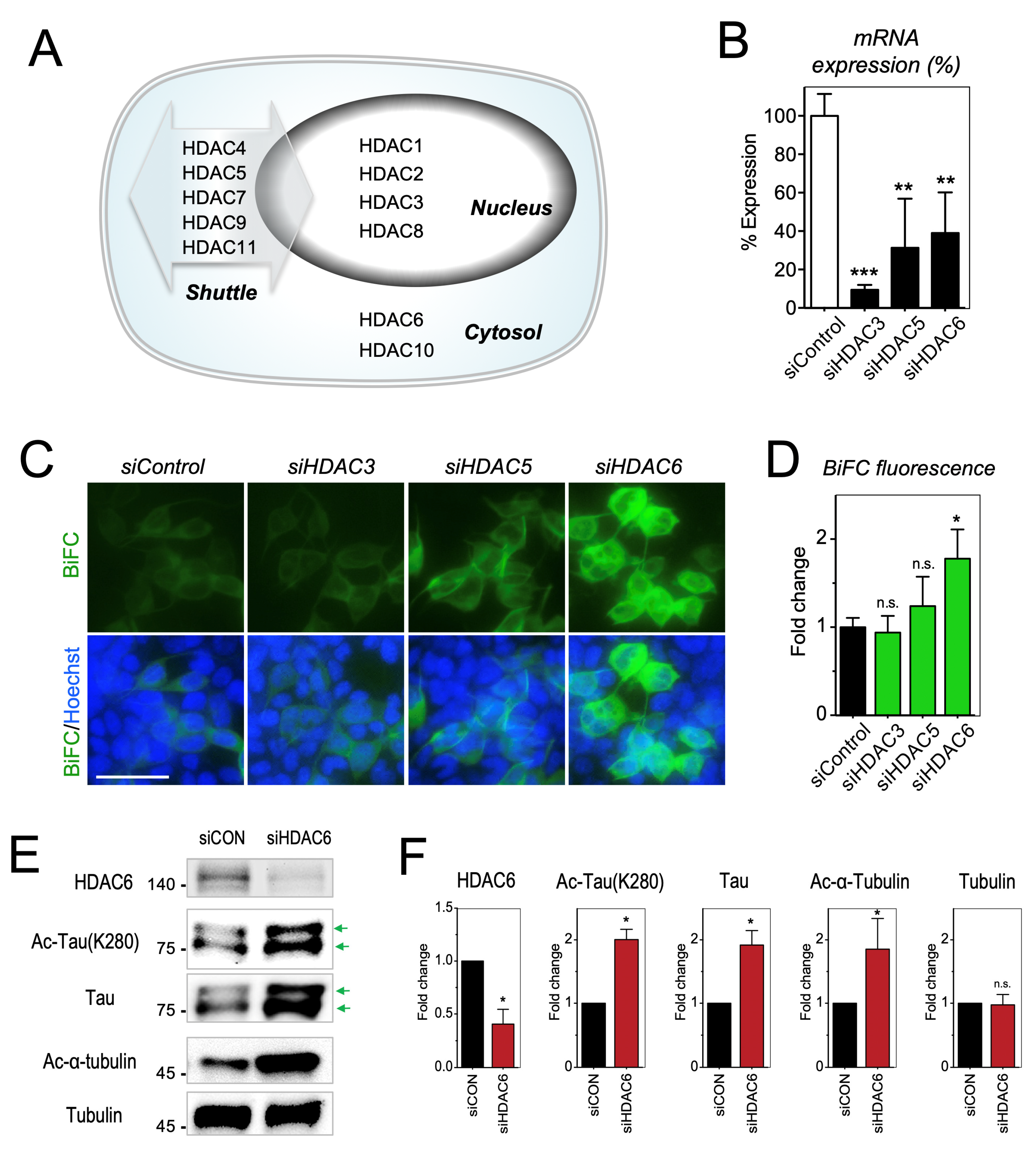
2.3. Increase of Tau Acetylation by the Inhibition of Cytoplasmic HDAC6
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Compound Screening
4.2. BiFC-Image Analysis
4.3. Immunoblot Analysis
4.4. Isolation of Mature Tau-BiFC Complexes Using GFP-Trap®
4.5. siRNA Transfection and Analysis
4.6. mRNA Extraction and Real-time Quantitative RT-PCR (QPCR) Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Murpy, M.; LeVine III, H. Alzheimer’s disease and the β-amyloid peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, H.; Jin, P. Epigenetics-based therapeutics for neurodegenerative disorders. Curr. Geriatr. Rep. 2012, 1, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.P.; LaFerla, F.M.; Oddo, S.S.; Brewer, G.J. Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer’s disease. Age 2013, 35, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Volmar, C.H.; Salah-Uddin, H.; Janczura, K.J.; Halley, P.; Lambert, G.; Wodrich, A.; Manoah, S.; Patel, N.H.; Sartor, G.C.; Mehta, N. M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing Alzheimer’s disease genes and improving memory. Proc. Natl. Acad. Sci. USA 2017, 114, E9135–E9144. [Google Scholar] [CrossRef] [PubMed]
- Francis, Y.I.; Fà, M.; Ashraf, H.; Zhang, H.; Staniszewski, A.; Latchman, D.S.; Arancio, O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2009, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.J.; Huang, F.I.; Liou, J.-P.; Yang, C.R. The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis. 2018, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Marco, S.; Pérez-Otaño, I.; García-Osta, A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 2012, 22, 1040–1050. [Google Scholar] [CrossRef]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef]
- Selenica, M.L.; Benner, L.; Housley, S.B.; Manchec, B.; Lee, D.C.; Nash, K.R.; Kalin, J.; Bergman, J.A.; Kozikowski, A.; Gordon, M.N. Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res. Ther. 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Ververis, K.; Karagiannis, T.C. Overview of the classical histone deacetylase enzymes and histone deacetylase inhibitors. ISRN Cell Biol. 2012, 2012. [Google Scholar] [CrossRef]
- Marks, P.A. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2010, 1799, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, H.; He, G.; Ly, P.T.; Fox, C.J.; Staufenbiel, M.; Cai, F.; Zhang, Z.; Wei, S.; Sun, X.; Chen, C.-H. Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J. Exp. Med. 2008, 205, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.P.; Xie, J.W.; Wang, C.Y.; Wang, T.; Wang, X.; Wang, S.L.; Teng, W.P.; Wang, Z.Y. Valproate reduces tau phosphorylation via cyclin-dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res. Bull. 2011, 85, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Pérez-Mediavilla, A.; Frechilla, D.; Del Río, J.; García-Osta, A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 2009, 34, 1721. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-L.; Yang, W.M. Beyond histone and deacetylase: An overview of cytoplasmic histone deacetylases and their nonhistone substrates. BioMed Res. Int. 2010, 2011. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef]
- Noack, M.; Leyk, J.; Richter-Landsberg, C. HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia 2014, 62, 535–547. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- PÎRŞCoveanu, D.F.V.; Pirici, I.; TudoricĂ, V.; BĂLŞEanu, T.A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; PÎRŞCoveanu, M. Tau protein in neurodegenerative diseases—A review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.Y. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimers Dis. 2008, 14, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Alonso, A.D.C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663. [Google Scholar] [CrossRef]
- Mazanetz, M.P.; Fischer, P.M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007, 6, 464. [Google Scholar] [CrossRef]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; Xie, S.X.; Lee, V.M.Y.; Trojanowski, J.Q. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 2012, 135, 807–818. [Google Scholar] [CrossRef]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; McCarty-Wood, E.; Van Deerlin, V.M.; Lee, V.M.-Y.; Trojanowski, J.Q. Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am. J. Pathol. 2013, 183, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Sohn, P.D.; Minami, S.S.; Wang, C.; Min, S.W.; Li, Y.; Zhou, Y.; Le, D.; Lo, I.; Ponnusamy, R. Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 2016, 90, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.A. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015, 21, 1154. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Kumari, A.; Panda, D. An acetylation mimicking mutation, K274Q, in tau imparts neurotoxicity by enhancing tau aggregation and inhibiting tubulin polymerization. Biochem. J. 2019, 476, 1401–1417. [Google Scholar] [CrossRef] [PubMed]
- Tak, H.; Haque, M.M.; Kim, M.J.; Lee, J.H.; Baik, J.-H.; Kim, Y.; Kim, D.J.; Grailhe, R.; Kim, Y.K. Bimolecular fluorescence complementation; lighting-up tau-tau interaction in living cells. PLoS ONE 2013, 8, e81682. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Haque, M.; Nam, G.; Ryoo, N.; Rhim, H.; Kim, Y. Monitoring of intracellular tau aggregation regulated by OGA/OGT inhibitors. Int. J. Mol. Sci. 2015, 16, 20212–20224. [Google Scholar] [CrossRef]
- Kim, D.; Lim, S.; Haque, M.M.; Ryoo, N.; Hong, H.S.; Rhim, H.; Lee, D.E.; Chang, Y.T.; Lee, J.S.; Cheong, E. Identification of disulfide cross-linked tau dimer responsible for tau propagation. Sci. Rep. 2015, 5, 15231. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Kim, D.; Ju, S.; Shin, S.; Cho, I.j.; Park, S.H.; Grailhe, R.; Lee, C.; Kim, Y.K. Glioblastoma-secreted soluble CD44 activates tau pathology in the brain. Exp. Mol. Med. 2018, 50, 5. [Google Scholar] [CrossRef]
- Shin, S.; Lim, S.; Jeong, H.; Kwan, L.; Kim, Y. Visualization of Tau–Tubulin Interaction in a Living Cell Using Bifluorescence Complementation Technique. Int. J. Mol. Sci. 2018, 19, 2978. [Google Scholar] [CrossRef]
- Liu, S.J.; Zhang, J.Y.; Li, H.L.; Fang, Z.Y.; Wang, Q.; Deng, H.M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K.; Wang, J.Z. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J. Biol. Chem. 2004, 279, 50078–50088. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, S.; CHENG, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.A.; Sun, Q.; Gamblin, T.C. Tau phosphorylation by GSK-3β promotes tangle-like filament morphology. Mol. Neurodegener. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Croucher, D.R.; Iconomou, M.; Hastings, J.F.; Kennedy, S.P.; Han, J.Z.; Shearer, R.F.; McKenna, J.; Wan, A.; Lau, J.; Aparicio, S. Bimolecular complementation affinity purification (BiCAP) reveals dimer-specific protein interactions for ERBB2 dimers. Sci. Signal. 2016, 9, ra69. [Google Scholar] [CrossRef] [PubMed]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A dual pathogenic mechanism links tau acetylation to sporadic tauopathy. Sci. Rep. 2017, 7, 44102. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Naushad, S.M.; Lavanya, A.; Srinivas, C.; Devi, T.A.; Sampathkumar, S.; Dharan, D.B.; Bhadra, M.P. Scriptaid cause histone deacetylase inhibition and cell cycle arrest in HeLa cancer cells: A study on structural and functional aspects. Gene 2017, 627, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Fleming, C.L.; Natoli, A.; Schreuders, J.; Devlin, M.; Yoganantharajah, P.; Gibert, Y.; Leslie, K.G.; New, E.J.; Ashton, T.D.; Pfeffer, F.M. Highly fluorescent and HDAC6 selective scriptaid analogues. Eur. J. Med. Chem. 2019, 162, 321–333. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Tapadar, S.; Luchini, D.N.; Kim, K.H.; Billadeau, D.D. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: A new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J. Med. Chem. 2008, 51, 4370–4373. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Qin, W.; Yi, F. Targeting histone deacetylases: Perspectives for epigenetic-based therapy in cardio-cerebrovascular disease. J. Geriatr. Cardiol. 2015, 12, 153. [Google Scholar]
- Lakshmaiah, K.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469. [Google Scholar] [PubMed]
- Carlomagno, Y.; Chung, D.E.C.; Yue, M.; Castanedes-Casey, M.; Madden, B.J.; Dunmore, J.; Tong, J.; DeTure, M.; Dickson, D.W.; Petrucelli, L. An acetylation–phosphorylation switch that regulates tau aggregation propensity and function. J. Biol. Chem. 2017, 292, 15277–15286. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.H.; Xie, L.; Song, S.; Xie, Y.; Allen, L.; Ajit, D.; Hong, J.S.; Chen, X.; Meeker, R.B.; Cohen, T.J. The Deacetylase HDAC6 mediates endogenous Neuritic tau pathology. Cell Rep. 2017, 20, 2169–2183. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone acetylation modifiers in the pathogenesis of Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Bahari-Javan, S.; Sananbenesi, F.; Fischer, A. Histone-acetylation: A link between Alzheimer’s disease and post-traumatic stress disorder? Front. Neurosci. 2014, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Rei, D.; Guan, J.-S.; Wang, W.-Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.; Fass, D.M.; Kao, P.F.; Kahn, M. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| HDAC Inhibitor | Target HDACs | References |
|---|---|---|
| Scriptaid | Class I (HDAC 1, 2, 3, 8) | [47,48] |
| Class IIb (HDAC 6) | ||
| M344 | Class I (HDAC 1, 2, 3, 8) | [6] |
| Class IIb (HDAC 6, 10) | ||
| BML281 | Class I (HDAC 3) | [49] |
| Class IIb (HDAC 6) | ||
| SAHA | Class I (HDAC 1, 2, 3, 8) | [50] |
| Class IIa (HDAC 4, 5, 7, 9) | ||
| Class IIb (HDAC 6, 10) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.; Shin, S.; Lee, J.-S.; Lee, S.H.; Baik, J.-H.; Lim, S.; Kim, Y.K. Pan-HDAC Inhibitors Promote Tau Aggregation by Increasing the Level of Acetylated Tau. Int. J. Mol. Sci. 2019, 20, 4283. https://doi.org/10.3390/ijms20174283
Jeong H, Shin S, Lee J-S, Lee SH, Baik J-H, Lim S, Kim YK. Pan-HDAC Inhibitors Promote Tau Aggregation by Increasing the Level of Acetylated Tau. International Journal of Molecular Sciences. 2019; 20(17):4283. https://doi.org/10.3390/ijms20174283
Chicago/Turabian StyleJeong, Hyeanjeong, Seulgi Shin, Jun-Seok Lee, Soo Hyun Lee, Ja-Hyun Baik, Sungsu Lim, and Yun Kyung Kim. 2019. "Pan-HDAC Inhibitors Promote Tau Aggregation by Increasing the Level of Acetylated Tau" International Journal of Molecular Sciences 20, no. 17: 4283. https://doi.org/10.3390/ijms20174283
APA StyleJeong, H., Shin, S., Lee, J. -S., Lee, S. H., Baik, J. -H., Lim, S., & Kim, Y. K. (2019). Pan-HDAC Inhibitors Promote Tau Aggregation by Increasing the Level of Acetylated Tau. International Journal of Molecular Sciences, 20(17), 4283. https://doi.org/10.3390/ijms20174283