Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation

 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Results
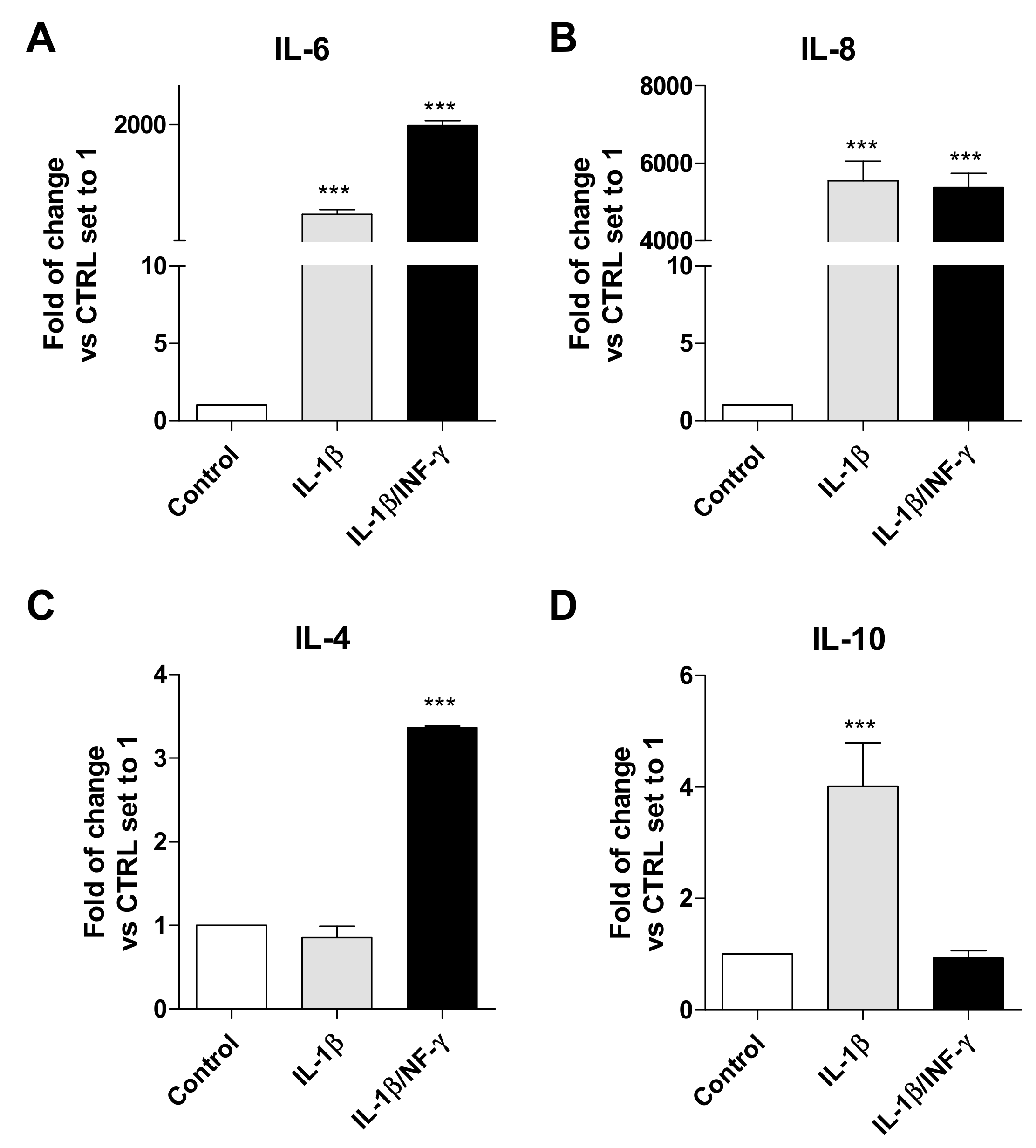
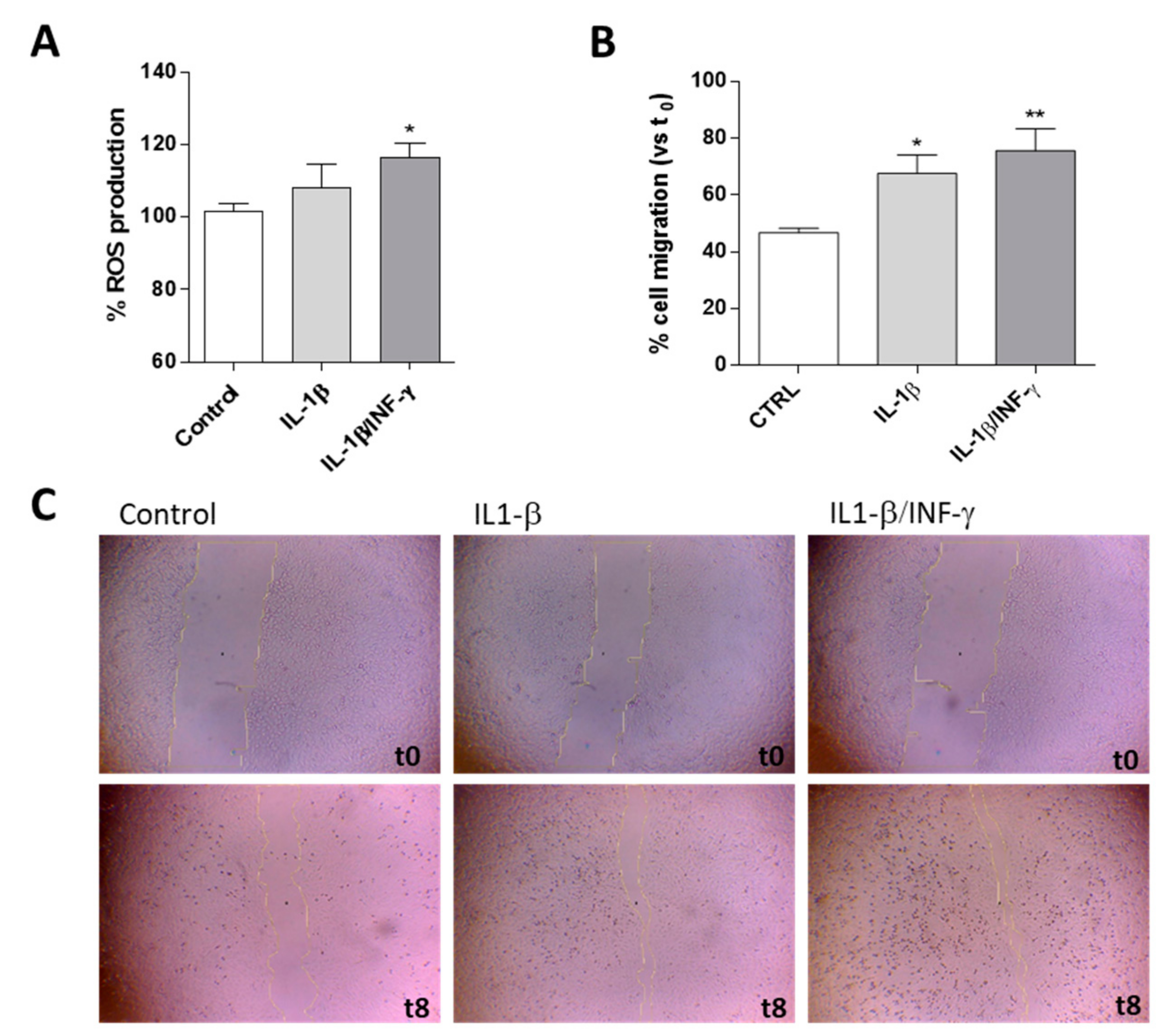
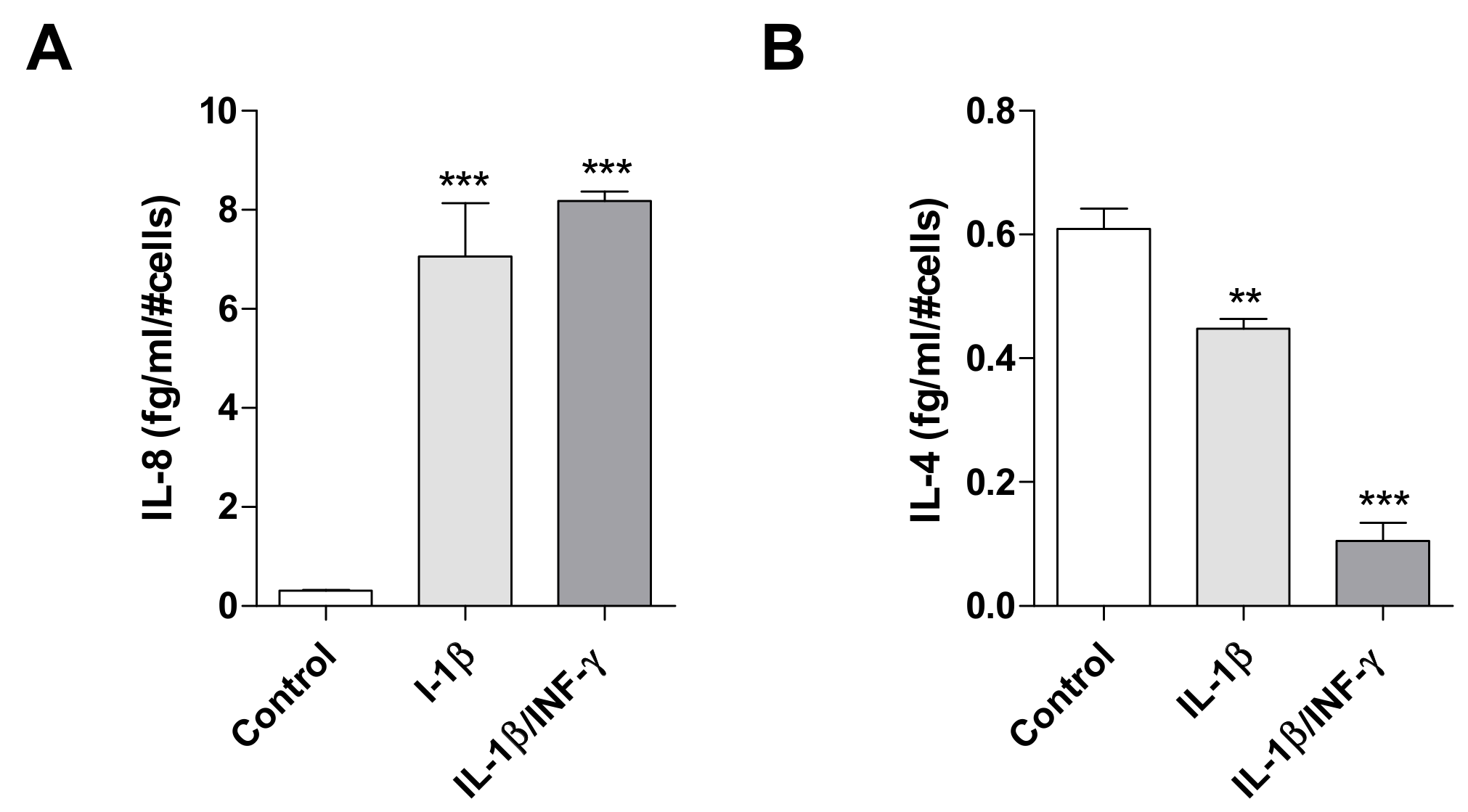
2.1. Human Microglial Inflammation: In Vitro Model Setting
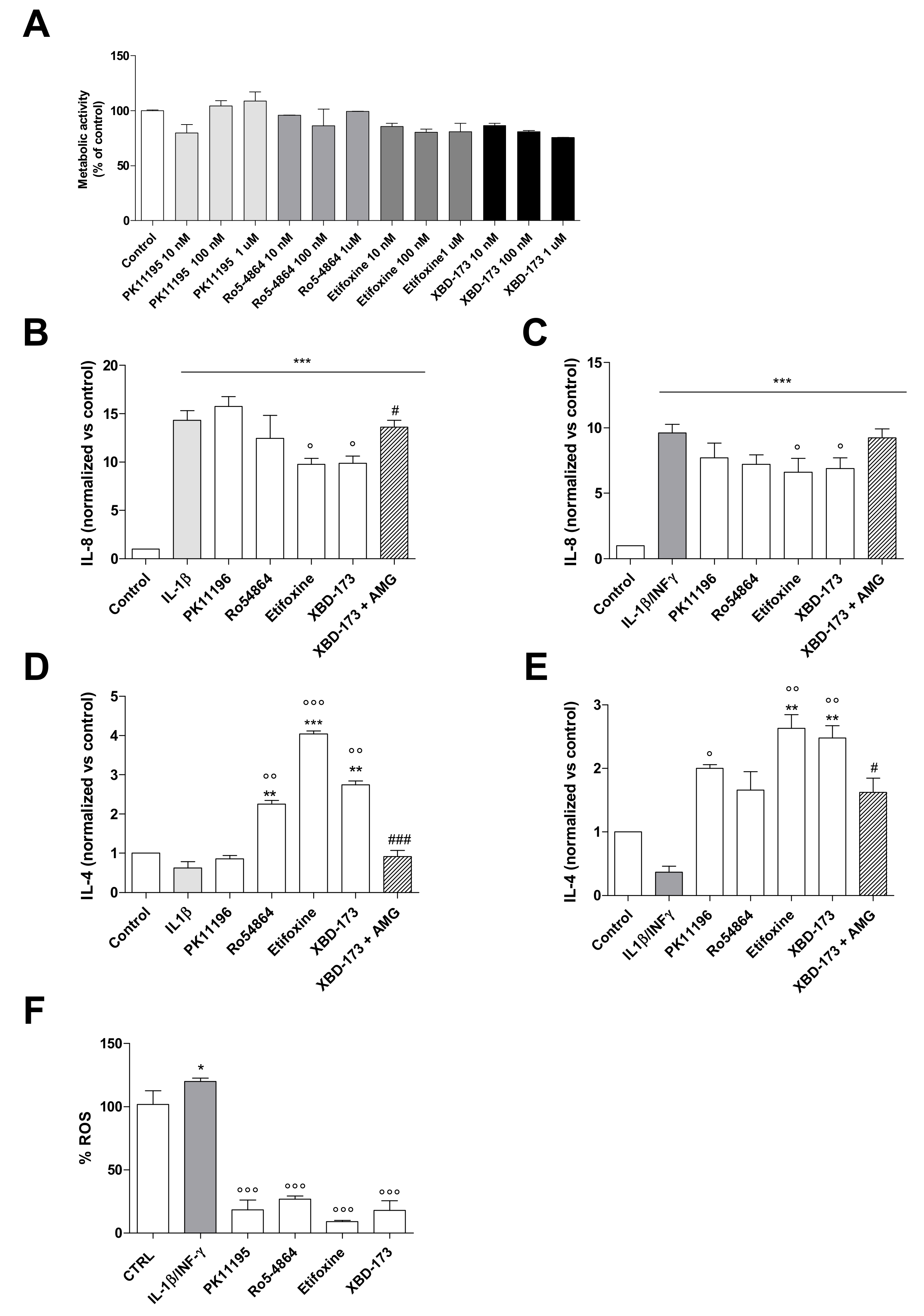
2.2. The Pharmacological Stimulation of TSPO Attenuates the Inflamed Phenotype of C20 Cells
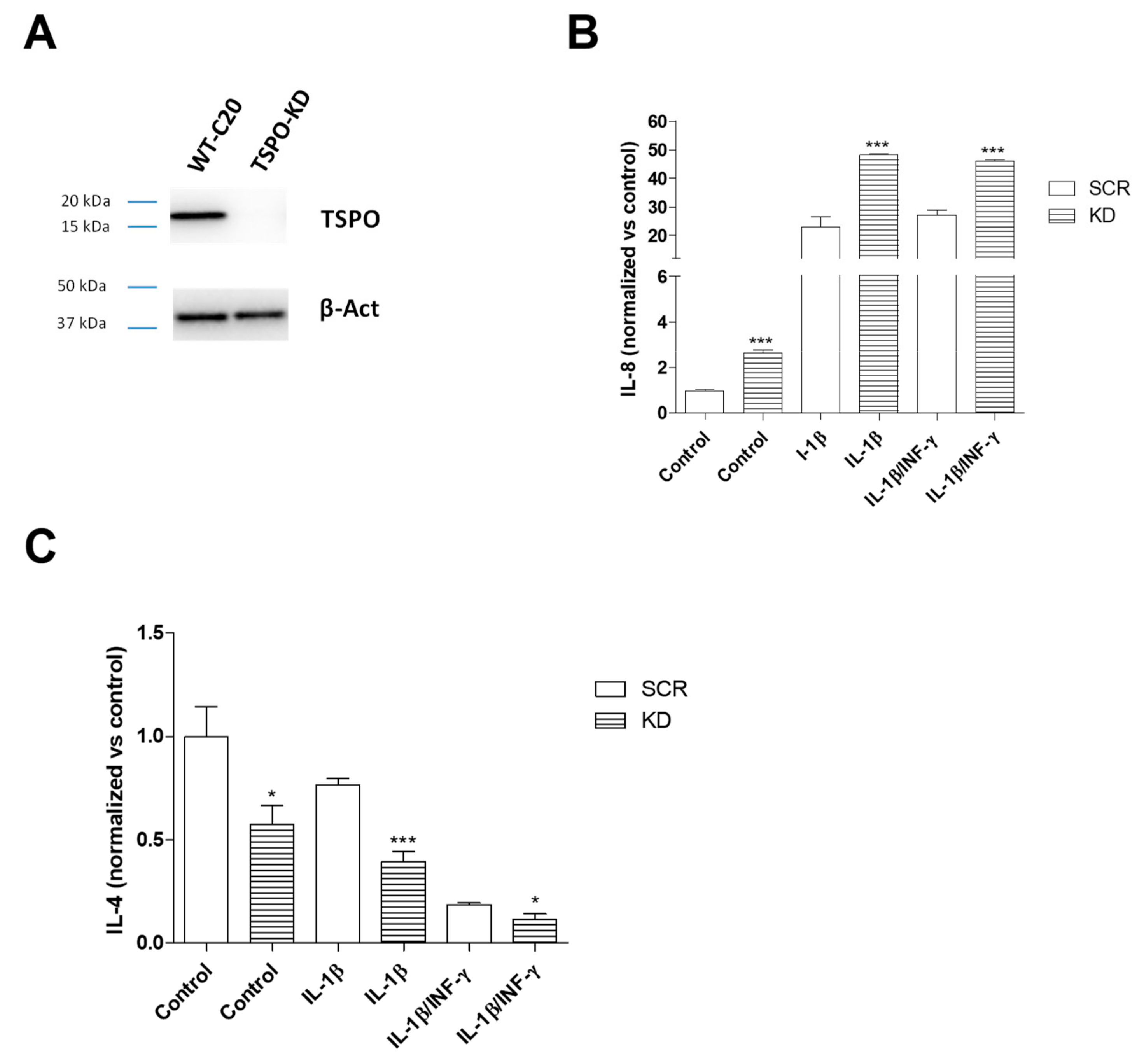
2.3. TSPO Knockdown Amplifies C20 Cell Responsiveness to the Inflammatory Stimulus
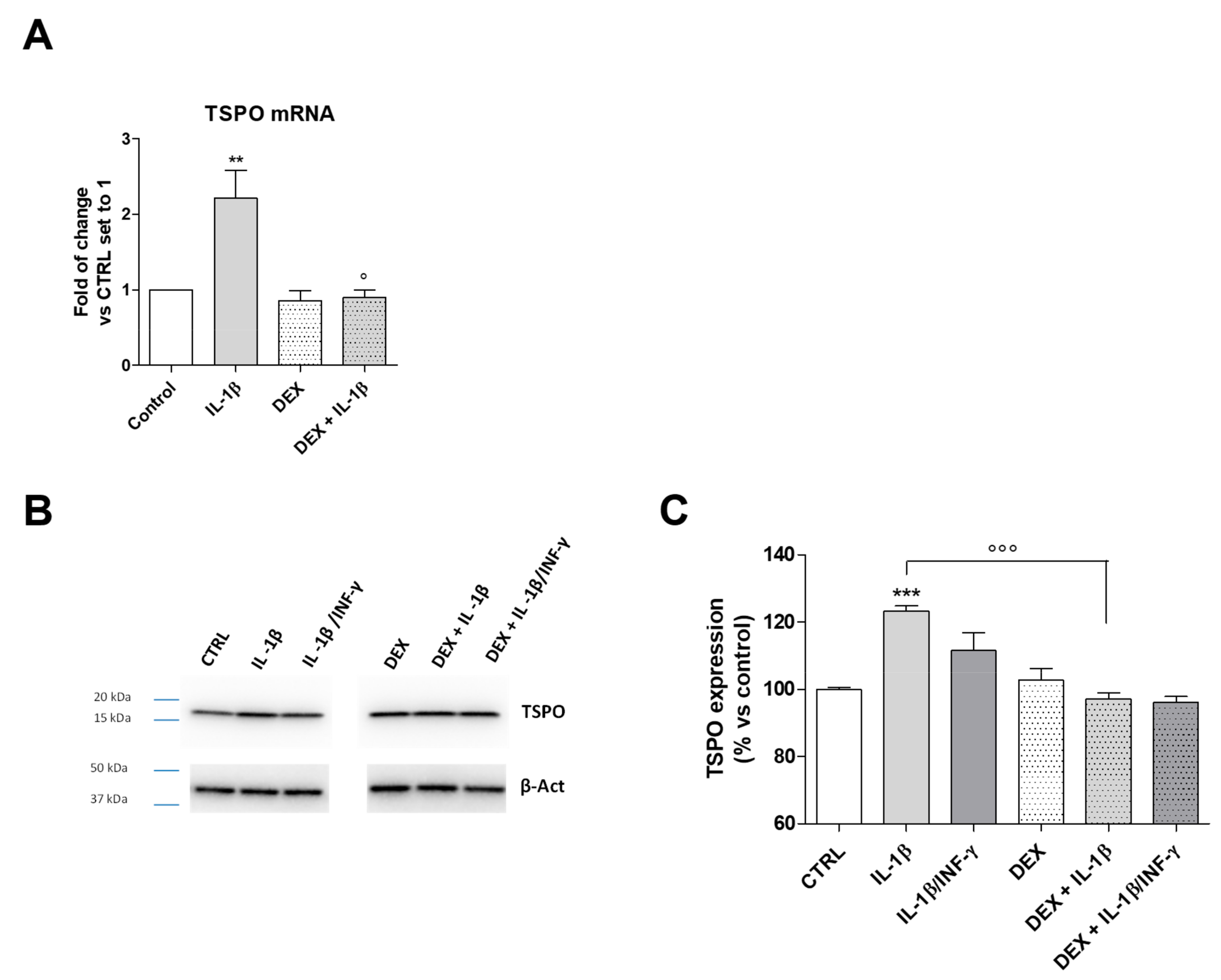
2.4. Regulation of TSPO Transcription in Microglia Following Inflammatory Stimuli
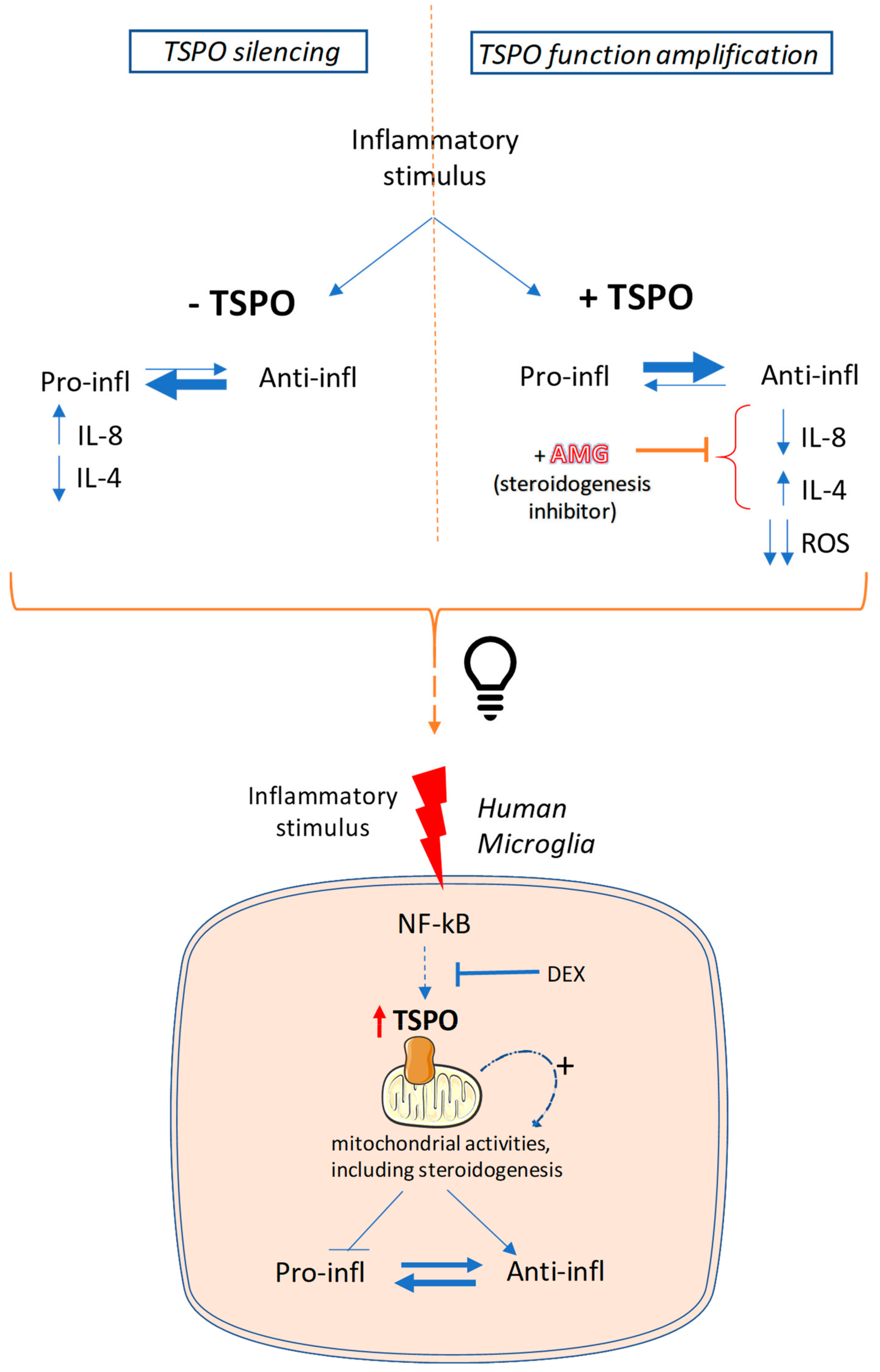
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Relative mRNA Quantification
4.4. ROS Production Evaluation
4.5. Western Blotting Analysis
4.6. Predictive Analysis of Transcription Factor Binding to the Human TSPO Promoter
4.7. Cell Viability Assay
4.8. Cytokine Production Quantification
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TSPO | Translocator Protein 18 KDa |
| KD | Knockdown |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| INF- γ | Interferon-γ |
| TNF-α | Tumor Necrosis Factor-α |
| VDAC | Voltage Dependent Anion Channel |
| CNS | Central Nervous System |
| CRAC | Cholesterol Recognition Aminoacidic Consensus |
| shRNA | Short Hairpin RNA |
| ROS | Reactive Oxygen Species |
| AMG | Aminoglutethimide |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| P450scc | Cytochrome P450 Side-Chain Cleavage |
| COX-2 | Cyclooxygenase-2 |
| TF | Transcription Factor |
| DEX | Dexamethasone |
| RE | Random Expectation |
| SCR | Scramble |
| TSS | Transcription Start Site |
References
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Fan, J.; Papadopoulos, V. Evolutionary origin of the mitochondrial cholesterol transport machinery revealsa universal mechanism of steroid hormone biosynthesis in animals. PLoS ONE 2013, 8, e76701. [Google Scholar] [CrossRef] [PubMed]
- Yasin, N.; Veenman, L.; Singh, S.; Azrad, M.; Bode, J.; Vainshtein, A.; Caballero, B.; Marek, I.; Gavish, M. Classical and Novel TSPO Ligands for the Mitochondrial TSPO Can Modulate Nuclear Gene Expression: Implications for Mitochondrial Retrograde Signaling. Int. J. Mol. Sci. 2017, 18, 786. [Google Scholar] [CrossRef]
- Lacor, P.; Gandolfo, P.; Tonon, M.C.; Brault, E.; Dalibert, I.; Schumacher, M.; Benavides, J.; Ferzaz, B. Regulation of the expression of peripheral benzodiazepine receptors and their endogenous ligands during rat sciatic nerve degeneration and regeneration: A role for PBR in neurosteroidogenesis. Brain Res. 1999, 815, 70–80. [Google Scholar] [CrossRef]
- Salter, M.; Stevens, S. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Fernàndez-Suàrez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Largeau, B.; Dupont, A.C.; Guilloteau, D.; Santiago-Ribeiro, M.J.; Arlicot, N. TSPO PET Imaging: From Microglial Activation to Peripheral Sterile Inflammatory Diseases? Contrast Media Mol. Imaging 2017, 2017, 6592139. [Google Scholar] [CrossRef]
- Arbo, B.D.; Benetti, F.; Garcia-Segura, L.M.; Ribeiro, M.F. Therapeutic actions of translocator protein (18 kDa) ligands in experimental models of psychiatric disorders and neurodegenerative diseases. J. Steroid Biochem. 2015, 154, 68–74. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018, 30. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Da Pozzo, E.; Martini, C. Translocator protein and steroidogenesis. Biochem. J. 2018, 475, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Da Pozzo, E.; Costa, B.; Martini, C. Translocator protein (TSPO) and neurosteroids: Implications in psychiatric disorders. Curr. Mol. Med. 2012, 12, 426–442. [Google Scholar] [PubMed]
- Gavish, M.; Veenman, L. Regulation of Mitochondrial, Cellular, and Organismal Functions by TSPO. Adv. Pharm. 2018, 82, 103–136. [Google Scholar]
- Ryu, J.K.; Choi, H.B.; McLarnon, J.G. Peripheral benzodiazepine receptor ligand PK11195 reduces microglial activation and neuronal death in quinolinic acid-injected rat striatum. Neurobiol. Dis. 2005, 20, 550–561. [Google Scholar] [CrossRef]
- Leaver, K.R.; Reynolds, A.; Bodard, S.; Guilloteau, D.; Chalon, S.; Kassiou, M. Effects of translocator protein (18 kDa) ligands on microglial activation and neuronal death in the quinolinic-acid-injected rat striatum. ACS Chem. Neurosci. 2012, 3, 114–119. [Google Scholar] [CrossRef]
- Ferzaz, B.; Brault, E.; Bourliaud, G.; Robert, J.P.; Poughon, G.; Claustre, Y.; Marguet, F.; Liere, P.; Schumacher, M.; Nowicki, J.P.; et al. SSR180575 (7-chloro-N,N,5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5-b]indole-1-acetamide), a peripheral benzodiazepine receptor ligand, promotes neuronal survival and repair. J. Pharmacol. Exp. Ther. 2002, 301, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Liu, S.; Cadepond, F.; Adams, D.; Lacroix, C.; Verleye, M.; Gillardin, J.M.; Baulieu, E.E.; Schumacher, M.; Schweizer-Groyer, G. Etifoxine improves peripheral nerve regeneration and functional recovery. Proc. Natl. Acad. Sci. USA 2008, 105, 20505–20510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga, S.; Azcoitia, I.; Garcia-Segura, L.M. Ro5-4864, a peripheral benzodiazepine receptor ligand, reduces reactive gliosis and protects hippocampal hilar neurons from kainic acid excitotoxicity. J. Neurosci. Res. 2005, 80, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga, S.; Carrero, P.; Pernia, O.; Azcoitia, I.; Garcia-Segura, L.M. Translocator protein 18 kDa is involved in the regulation of reactive gliosis. Glia 2007, 55, 1426–1436. [Google Scholar] [CrossRef]
- Choi, J.; Ifuku, M.; Noda, M.; Guilarte, T.R. Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia 2011, 59, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; da Pozzo, E.; Giacomelli, C.; Barresi, E.; Taliani, S.; da Settimo, F.; Martini, C. TSPO ligand residence time: A new parameter to predict compound neurosteroidogenic efficacy. Sci. Rep. 2016, 6, 18164. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Cavallini, C.; Da Pozzo, E.; Taliani, S.; Da Settimo, F.; Martini, C. The Anxiolytic Etifoxine Binds to TSPO Ro5-4864 Binding Site with Long Residence Time Showing a High Neurosteroidogenic Activity. ACS Chem. Neurosci. 2017, 8, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Taliani, S.; Da Pozzo, E.; Barresi, E.; Robello, M.; Cavallini, C.; Cosconati, S.; da Settimo, F.; Novellino, E.; Martini, C. Residence Time, a New parameter to Predict Neurosteroidogenic Efficacy of Translocator Protein (TSPO) Ligands: The Case Study of N,N-Dialkyl-2-arylindol-3-ylglyoxylamides. ChemMedChem 2017, 12, 1275–1278. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; da Pozzo, E.; Cavallini, C.; Taliani, S.; Da Settimo, F.; Martini, C. Long Residence Time at the Neurosteroidogenic 18 kDa Translocator Protein Characterizes the Anxiolytic Ligand XBD173. ACS Chem. Neurosci. 2016, 7, 1041–1046. [Google Scholar] [CrossRef]
- Da Pozzo, E.; Giacomelli, C.; Costa, B.; Cavallini, C.; Taliani, S.; Barresi, E.; da Settimo, F.; Martini, C. TSPO PIGA Ligands Promote Neurosteroidogenesis and Human Astrocyte Well-Being. Int. J. Mol. Sci. 2016, 17, 1028. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Mattace Raso, G.; Taliani, S.; da Pozzo, E.; Simorini, F.; Costa, B.; Martini, C.; Laneri, S.; Sacchi, A.; Cosimelli, B.; et al. TSPO-ligands prevent oxidative damage and inflammatory response in C6 glioma cells by neurosteroid synthesis. Eur. J. Pharm. Sci. 2016, 88, 124–131. [Google Scholar] [CrossRef]
- Smith, A.M.; Dragunow, M. The human side of microglia. Trends Neurosci. 2014, 37, 125–135. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Jay, T.R.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017, 23, 47–66. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ + TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Davis, R.L.; Buck, D.J.; McCracken, K.; Cox, G.W.; Das, S. Interleukin-1β-induced inflammatory signaling in C20 human microglial cells. Neuroimmunol. Neuroinflamm. 2018, 5, 50. [Google Scholar] [CrossRef]
- Lee, S.C.; Liu, W.; Dickson, D.W.; Brosnan, C.F.; Berman, J.W. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 β. J. Immunol. 1993, 150, 2659–2667. [Google Scholar] [PubMed]
- Kaushik, D.K.; Thounaojam, M.C.; Kumawat, K.L.; Gupta, M.; Basu, A. Interleukin-1β orchestrates underlying inflammatory responses in microglia via Krüppel-like factor 4. J. Neurochem. 2013, 127, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflamm. 2018, 15, 259. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Park, T.I.; Schweder, P.; Scotter, J.; Correia, J.; Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; et al. Isolation of highly enriched primary human microglia for functional studies. Sci. Rep. 2016, 18, 19371. [Google Scholar] [CrossRef]
- Da Pozzo, E.; Giacomelli, C.; Cavallini, C.; Martini, C. Cytokine secretion responsiveness of lymphomonocytes following cortisol cell exposure: Sex differences. PLoS ONE 2018, 13, e0200924. [Google Scholar] [CrossRef]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef]
- Karlstetter, M.; Nothdurfter, C.; Aslanidis, A.; Moeller, K.; Horn, F.; Scholz, R.; Neumann, H.; Weber, B.H.; Rupprecht, R.; Langmann, T. Translocator protein (18 kDa) (TSPO) is expressed in reactive retinal microglia and modulates microglial inflammation and phagocytosis. J. Neuroinflamm. 2014, 11, 3. [Google Scholar] [CrossRef]
- Fan, Y.; Xie, L.; Chung, C.Y. Signaling Pathways Controlling Microglia Chemotaxis. Mol. Cells 2017, 40, 163–168. [Google Scholar] [CrossRef]
- Derecki, N.C.; Cardani, A.N.; Yang, C.H.; Quinnies, K.M.; Crihfield, A.; Lynch, K.R.; Kipnis, J. Regulation of learning and memory by meningeal immunity: A key role for IL-4. J. Exp. Med. 2010, 207, 1067–1080. [Google Scholar] [CrossRef]
- Ehrlich, L.C.; Shuxian, H.; Sheng, W.S.; Sutton, R.L.; Rockswold, G.L.; Peterson, P.K.; Chao, C.C. Cytokine Regulation of Human Microglial Cell IL-8 Production. J. Immunol. 1998, 160, 1944–1948. [Google Scholar] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, F.; Lamarche, I.; Prabonnaud, V.; le Fur, G.; Herbert, J.M. Peripheral Benzodiazepine Receptor Agonists Exhibit Potent Antiapoptotic Activities. Biochem. Biophys. Res. Commun. 1999, 265, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Strohmeier, R.; Roller, M.; Sänger, N.; Knecht, R.; Kuhl, H. Modulation of tamoxifen-induced apoptosis by peripheral benzodiazepine receptor ligands in breast cancer cells. Biochem. Pharmacol. 2002, 64, 99–107. [Google Scholar] [CrossRef]
- Leducq, N.; Bono, F.; Sulpice, T.; Vin, V.; Janiak, P.; Fur, G.L.; O’Connor, S.E.; Herbert, J.M. Role of peripheral benzodiazepine receptors in mitochondrial, cellular, and cardiac damage induced by oxidative stress and ischemia-reperfusion. J. Pharmacol. Exp. Ther. 2003, 306, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Hamon, A.; Morel, A.; Hue, B.; Verleye, M.; Gillardin, J.M. The modulatory effects of the anxiolytic Etifoxine on GABA (A) receptors are mediated by the beta subunit. Neuropharmacology 2003, 45, 293–303. [Google Scholar] [CrossRef]
- Bader, S.; Wolf, L.; Milenkovic, V.; Gruber, M.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuro 2019, 106, 65–76. [Google Scholar] [CrossRef]
- Wang, M.; Wang, X.; Zhao, L.; Ma, W.; Rodriguez, I.R.; Fariss, R.N.; Wong, W.T. Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. J. Neurosci. 2014, 34, 3793–3806. [Google Scholar] [CrossRef]
- Gatliff, J.; East, D.A.; Singh, A.; Alvarez, M.S.; Frison, M.; Matic, I.; Ferraina, C.; Sampson, N.; Turkheimer, F.; Campanella, M. A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis. 2017, 22, e2896. [Google Scholar] [CrossRef]
- Strobner, P.E.; Carayon, P.; Casellas, P.; Portier, M.; Lavabre-Bertrand, T.; Cuq, P.; Cano, J.P.; Meynadier, J.; Meunier, L. Transient protection by peripheral benzodiazepine receptors during the early events of ultraviolet light-induced apoptosis. Cell Death Differ. 2001, 8, 747–753. [Google Scholar] [CrossRef]
- Ratcliffe, S.L.; Matthews, E.K. Modification of the photodynamic action of δ-aminolaevulinic acid (ALA) on rat pancreatoma cells by mitochondrial benzodiazepine receptor ligands. Br. J. Cancer 1995, 71, 300–305. [Google Scholar] [CrossRef]
- Choi, H.B.; Khoo, C.; Ryu, J.K.; van Breemen, E.; Kim, S.U.; McLarnon, J.G. Inhibition of lipopolysaccharide-induced cyclooxygenase-2, tumor necrosis factor-α and [Ca2+]i responses in human microglia by the peripheral benzodiazepine receptor ligand PK11195. J. Neurochem. 2002, 83, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Choi, H.B.; Kim, S.U.; McLarnon, J.G. Mitochondrial ligand inhibits store-operated calcium influx and COX-2 production in human microglia. J. Neurosci. Res. 2006, 83, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Carayon, P.; Portier, M.; Dussossoy, D.; Bord, A.; Petitprêtre, G.; Canat, X.; le Fur, G.; Casellas, P. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood 1996, 87, 3170–3178. [Google Scholar] [PubMed]
- Veenman, L.; Shandalov, Y.; Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 2008, 40, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Sutter, A.P.; Maaser, K.; Höpfner, M.; Barthel, B.; Grabowski, P.; Faiss, S.; Carayon, P.; Zeitz, M.; Scherübl, H. Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human esophageal cancer cells. Int. J. Cancer 2002, 102, 318–327. [Google Scholar] [CrossRef]
- Chelli, B.; Lena, A.; Vanacore, R.; da Pozzo, E.; Costa, B.; Rossi, L.; Salvetti, A.; Scatena, F.; Ceruti, S.; Abbracchio, M.P.; et al. Peripheral benzodiazepine receptor ligands: Mitochondrial transmembrane potential depolarization and apoptosis induction in rat C6 glioma cells. Biochem. Pharmacol. 2004, 68, 125–134. [Google Scholar] [CrossRef]
- Chelli, B.; Rossi, L.; Da Pozzo, E.; Costa, B.; Spinetti, F.; Rechichi, M.; Salvetti, A.; Lena, A.; Simorini, F.; Vanacore, R.; et al. PIGA (N,N-Di-n-butyl-5-chloro-2-(4-chlorophenyl)indol-3-ylglyoxylamide), a new mitochondrial benzodiazepine-receptor ligand, induces apoptosis in C6 glioma cells. Chembiochem 2005, 6, 1082–1088. [Google Scholar] [CrossRef]
- Chelli, B.; Salvetti, A.; Da Pozzo, E.; Rechichi, M.; Spinetti, F.; Rossi, L.; Costa, B.; Lena, A.; Rainaldi, G.; Scatena, F.; et al. PK11195 differentially affects cell survival in human wild-type and 18kDa translocator protein silenced ADF astrocytoma cells. J. Cell. Biochem. 2008, 105, 712–723. [Google Scholar] [CrossRef]
- Batarseh, A.; Barlow, K.D.; Martinez-Arguelles, D.B.; Papadopoulos, V. Functional characterization of the human translocator protein (18 kDa) gene promoter in human breast cancer cell lines. Biochim. Biophys. Acta 2012, 1819, 38–56. [Google Scholar] [CrossRef]
- Crinelli, R.; Antonelli, A.; Bianchi, M.; Gentilini, L.; Scaramucci, S.; Magnani, M. Selective inhibition of NF-κB activation and TNF-α production in macrophages by red blood cell-mediated delivery of dexamethasone. Blood Cells Mol. Dis. 2000, 26, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Baldanta, S.; Fernández-Escobar, M.; Acín-Perez, R.; Albert, M.; Camafeita, E.; Jorge, I.; Vázquez, J.; Enríquez, J.A.; Guerra, S. ISG15 governs mitochondrial function in macrophages following vaccinia virus infection. PLoS Pathog. 2017, 13, e1006651. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Eggen, B.J.; Raj, D.; Hanisch, U.K.; Boddeke, H.W. Microglial phenotype and adaptation. J. Neuroimmune Pharmacol. 2013, 8, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Lanussa, O.H.; Ávila-Rodriguez, M.; Garcìa-Segura, L.M.; González, J.; Echeverria, V.; Aliev, G.; Barreto, G.E. Microglial dependent protective effects of neuroactive steroids. CNS Neurol. Disord. Drug Targets 2016, 15, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Giatti, S.; Boraso, M.; Melcangi, R.C.; Viviani, B. Neuroactive steroids, their metabolites, and neuroinflammation. J. Mol. Endocrinol. 2012, 49, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Scholz, R.; Caramoy, A.; Bhuckory, M.B.; Rashid, K.; Chen, M.; Xu, H.; Langmann, T. Targeting translocator protein (18 kDa) (TSPO) dampens pro-inflammatory microglia reactivity in the retina and protects from degeneration. J. Neuroinflamm. 2015, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br. J. Pharmacol. 2006, 148, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Leak, R.K.; Hu, X. Neurotransmitter receptors on microglia. Stroke Vasc. Neurol. 2016, 1, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Schwab, C.; McGeer, P.L. Astrocytes are GABAergic cells that modulate microglial activity. Glia 2011, 59, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.R.; Shim, H.J.; Balu, D.; Kim, S.R.; Yu, S.W. Translocator protein 18 kDa negatively regulates inflammation in microglia. J. Neuroimmune Pharmacol. 2014, 9, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, S.; Wang, W.; Cao, Z.; Dan, J.; Cheng, J.; Cao, W.; Tian, F.; Cao, W.; Tian, Y. Potential involvement of the 18 kDa translocator protein and reactive oxygen species in apoptosis of THP-1 macrophages induced by sonodynamic therapy. PLoS ONE 2018, 13, e0196541. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Berkovich, A.; Krueger, K.E.; Costa, E.; Guidotti, A. Diazepam binding inhibitor and its processing products stimulate mitochondrial steroid biosynthesis via an interaction with mitochondrial benzodiazepine receptors. Endocrinology 1991, 129, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.J.; Kam, W.W.Y.; Liu, G.J.; Banati, R.B. Epigenetic Silencing of the Human 18 kDa Translocator Protein in a T Cell Leukemia Cell Line. DNA Cell Biol. 2017, 36, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.; Grueneberg, D.A.; Yang, X.; Kim, S.Y.; Kloepfer, A.M.; Hinkle, G.; Piqani, B.; Eisenhaure, T.M.; Luo, B.; Grenier, J.K.; et al. A Lentiviral RNAi Library for Human and Mouse Genes Applied to an Arrayed Viral High-Content Screen. Cell 2006, 124, 1283–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef] [Green Version]
- Dunigan, D.D.; Waters, S.B.; Owen, T.C. Aqueous soluble tetrazolium/formazan MTS as an indicator of NADH- and NADPH-dependent dehydrogenase activity. BioTechniques 1995, 19, 640–649. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Nucleotide Sequences | Product Size (Base Pair) | Annealing Temperature |
|---|---|---|---|
| IL-6 | FOR: 5′-TCCTCGACGGCATCTCA-3′ REV: 5′-TTTTCACCAGGCAAGTCTCCT-3′ | 165 bp | 55°C |
| IL-8 | FOR: 5′-AAGAGAGCTCTGTCTGGACC-3′ REV: 5′-GATATTCTCTTGGCCCTTGG-3′ | 408 bp | 56°C |
| IL-4 | FOR: 5′-ACTTTGAACAGCCTCACAGAG-3′ REV: 5′-TTGGAGGCAGCAAAGATGTC-3′ | 74 bp | 56°C |
| IL-10 | FOR: 5′-CAAGCTGAGAACCAAGACCC-3′ REV: 5′-AAGATGTCAAACTCACTCATGGC-3′ | 141 bp | 55°C |
| TSPO | FOR: 5′-CTTTGGTGCCCGACAAATGG-3′ REV:5′-CTGACCAGCAGGAGATCCAC-3′ | 51 bp | 55°C |
| β-actin | FOR: 5′-GCACTCTTCCAGCCTTCCTTCC-3′ REV: 5′-GAGCCGCCGATCCACACG-3′ | 254 bp | 55°C |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Pozzo, E.; Tremolanti, C.; Costa, B.; Giacomelli, C.; Milenkovic, V.M.; Bader, S.; Wetzel, C.H.; Rupprecht, R.; Taliani, S.; Da Settimo, F.; et al. Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation. Int. J. Mol. Sci. 2019, 20, 4467. https://doi.org/10.3390/ijms20184467
Da Pozzo E, Tremolanti C, Costa B, Giacomelli C, Milenkovic VM, Bader S, Wetzel CH, Rupprecht R, Taliani S, Da Settimo F, et al. Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation. International Journal of Molecular Sciences. 2019; 20(18):4467. https://doi.org/10.3390/ijms20184467
Chicago/Turabian StyleDa Pozzo, Eleonora, Chiara Tremolanti, Barbara Costa, Chiara Giacomelli, Vladimir M. Milenkovic, Stefanie Bader, Christian H. Wetzel, Rainer Rupprecht, Sabrina Taliani, Federico Da Settimo, and et al. 2019. "Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation" International Journal of Molecular Sciences 20, no. 18: 4467. https://doi.org/10.3390/ijms20184467
APA StyleDa Pozzo, E., Tremolanti, C., Costa, B., Giacomelli, C., Milenkovic, V. M., Bader, S., Wetzel, C. H., Rupprecht, R., Taliani, S., Da Settimo, F., & Martini, C. (2019). Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation. International Journal of Molecular Sciences, 20(18), 4467. https://doi.org/10.3390/ijms20184467