Comparative Analysis of Proteomics and Transcriptomics during Fertility Transition in a Two-Line Hybrid Rice Line Wuxiang S


Abstract
:
1. Introduction
2. Results
2.1. Cytological Characteristics of WXS during Fertility Transition
2.2. Overview of Quantitative Proteomics Analysis
2.3. Transcriptional Analysis of Differentially Expressed Genes (DEGs)
2.4. Comparison Analysis Between the Transcriptome and Proteome Data
2.5. GO and Pathway Enrichment Analysis of the cor-DEGs-DAPs Genes
2.6. The cor-DEGs-DAPs Genes Enriched in the Cell Wall of the GO Term
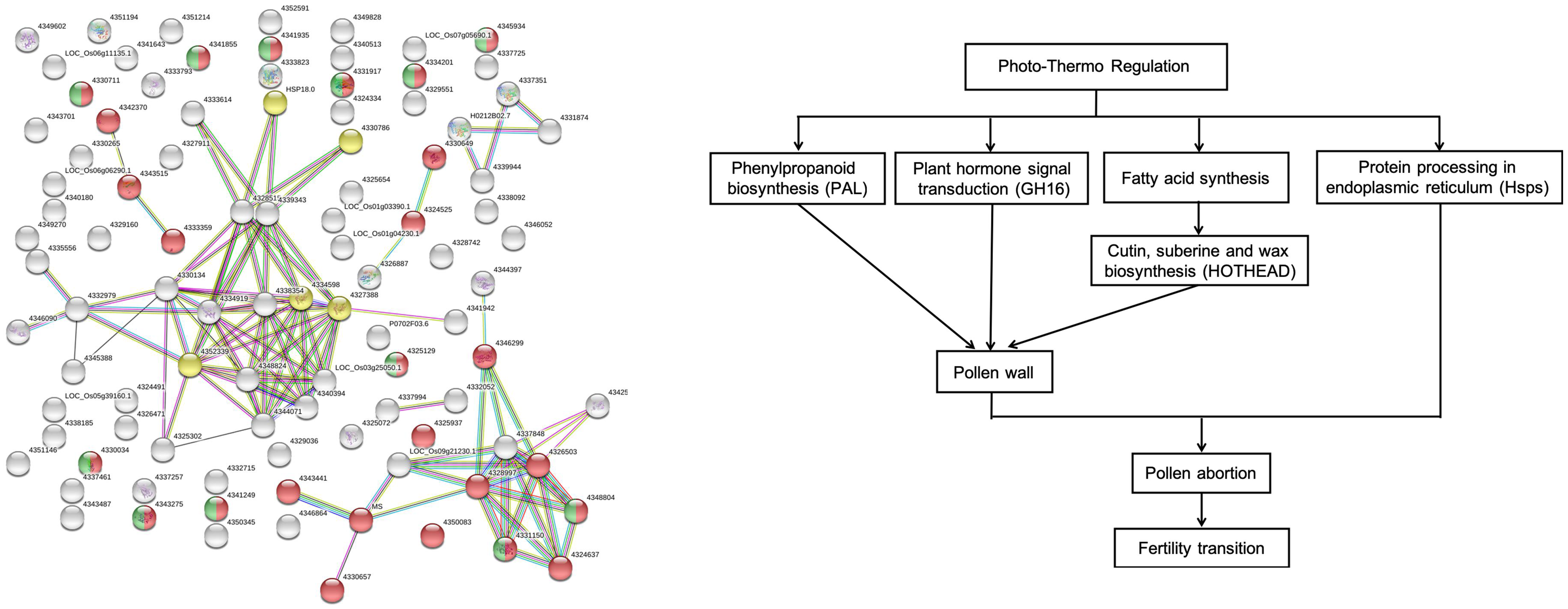
2.7. The Protein–Protein Interaction Network of the cor-DEGs-DAPs Proteins
2.8. The cor-DEGs-DAPs Genes Examined by qPCR
3. Discussion
3.1. Plant Hormone Signal Transduction
3.2. Protein Processing in Endoplasmic Reticulum
3.3. Starch and Sucrose Metabolism
3.4. Phenylpropanoid Biosynthesis
3.5. Cutin, Suberine, and Wax Biosynthesis
3.6. Proposed Model for Gene Transcript Regulation of PTGMS Rice during the Fertility Transition Stage
4. Materials and Methods
4.1. Plant Materials
4.2. Protein Preparation
4.3. Protein Digestion
4.4. iTRAQ Labeling
4.5. High pH Reverse Phase Fractionation
4.6. RPLC-ESI-MS/MS Analysis Based on Triple TOF 5600
4.7. iTRAQ Data Analysis
4.8. Protein Annotation and Classification
4.9. Protein Interaction Analysis Using STRING
4.10. RNA-seq and Data Analysis
4.11. RNA Isolation and Real Time RT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PTGMS | photo-thermosensitive genic male sterile |
| DEP | differentially expressed gene |
| DAP | differentially abundant protein |
| cor-DEGs-DAPs | corresponding DEGs and DAPs |
| qRT-PCR | quantitative RT-PCR |
| PMC | pollen mother cell |
| P2 | the PMC formation stage |
| P3 | the meiosis stage of PMC |
References
- Sheng, Z.H.; Wei, X.J.; Shao, G.N.; Chen, M.L.; Song, J.; Tang, S.Q.; Luo, J.; Hu, Y.C.; Hu, P.S.; Chen, L.Y. Genetic analysis and fine mapping of tms9, a novel thermosensitive genic male-sterile gene in rice (Oryza sativa L.). Plant Breed 2013, 132, 159–164. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, Q. Genetic and molecular characterization of photoperiod and thermo-sensitive male sterility in rice. Plant Reprod. 2017, 31, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zhang, Z.; Wu, B.; Chen, X.; Zhang, G.; Zhang, Z.; Wan, B.; Lu, Y. Molecular mapping of two reverse photoperiod-sensitive genic male sterility genes (rpms1 and rpms2) in rice (Oryza sativa L.). Theor. Appl. Genet. 2008, 118, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.K.; Liang, C.Y.; Zhuang, W.; Li, J.; Deng, H.B.; Deng, Q.Y.; Wang, B. Characterization and identification of the candidate gene of rice thermo-sensitive genic male sterile gene tms5 by mapping. Planta 2007, 225, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Q.; Li, J.; Jiang, D.; Zhou, L.; Wu, P.; Lu, S.; Li, F.; Zhu, L.; Liu, Z.; et al. Photoperiod- and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res. 2012, 22, 649–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, B.; Wu, Y.; Du, P.; Wang, J.; Wang, M.; Yi, C.; Gu, M.; Liang, G. Fine mapping and candidate gene analysis of ptgms2-1, the photoperiod-thermo-sensitive genic male sterile gene in rice (Oryza sativa L.). Theor. Appl. Genet. 2011, 122, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Nas, T.M.S.; Sanchez, D.L.; Diaz, G.Q.; Mendioro, M.S.; Virmani, S.S. Pyramiding of thermosensitive genetic male sterility (TGMS) genes and identification of a candidate tms5 gene in rice. Euphytica 2005, 145, 67–75. [Google Scholar] [CrossRef]
- MS, S. The discovery and study of the photosensitive recessive male sterile rice. Sci. Agric. Sin. 1985, 2, 44–48. (In Chinese) [Google Scholar]
- Shi, Y.; Zhao, S.; Yao, J. Premature tapetum degeneration: A major cause of abortive pollen development in photoperiod sensitive genic male sterility in rice. J. Integr. Plant. Biol. 2009, 51, 774–781. [Google Scholar] [CrossRef]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, M.; Yang, Y.; Li, J.; Zhu, L.; Jiang, D.; Dong, J.; Liu, Q.; Gu, L.; Zhou, L.; et al. RNase Z(S1) processes UbL40 mRNAs and controls thermosensitive genic male sterility in rice. Nat. Commun. 2014, 5, 4884. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yang, Y.; Yang, Y.; Lin, J.; Tang, D.; Liu, X. Comparative analysis of young panicle proteome in thermo-sensitive genic male-sterile rice Zhu-1S under sterile and fertile conditions. Biotechnol. Lett. 2009, 31, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pang, C.; Wei, H.; Song, M.; Meng, Y.; Fan, S.; Yu, S. Proteomic analysis of anthers from wild-type and photosensitive genetic male sterile mutant cotton (Gossypium hirsutum L.). BMC Plant. Biol. 2014, 14, 390. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, I.S.; Ross, A.R.; Olson, D.J.; Sawhney, V.K. Differential expression of proteins in the wild type and 7B-1 male-sterile mutant anthers of tomato (Solanum lycopersicum): A proteomic analysis. J. Proteomics 2009, 71, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Liu, Z.; Tong, J.; Xiao, L.; Ma, H.; Zhang, H. Comparative proteomics analysis reveals the mechanism of fertility alternation of thermosensitive genic male sterile rice lines under low temperature inducement. Proteomics 2015, 15, 1884–1905. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pang, C.; Wei, H.; Song, M.; Meng, Y.; Ma, J.; Fan, S.; Yu, S. Data for proteomic profiling of Anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). Data Brief. 2015, 4, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Zieske, L.R. A perspective on the use of iTRAQ (TM) reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Frost, D.C.; Greer, T.; Xiang, F.; Liang, Z.D.; Li, L.J. Development and characterization of novel 8-plex DiLeu isobaric labels for quantitative proteomics and peptidomics. Rapid Commun. Mass Spectrom. 2015, 29, 1115–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minic, Z. Physiological roles of plant glycoside hydrolases. Planta 2008, 227, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Opassiri, R.; Pomthong, B.; Onkoksoong, T.; Akiyama, T.; Esen, A.; Ketudat Cairns, J.R. Analysis of rice glycosyl hydrolase family 1 and expression of Os4bglu12 beta-glucosidase. BMC Plant. Biol. 2006, 6, 33. [Google Scholar] [CrossRef]
- Honys, D.; Twell, D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004, 5, R85. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Ischebeck, T.; Egelhofer, V.; Lichtscheidl, I.; Weckwerth, W. Cell-specific analysis of the tomato pollen proteome from pollen mother cell to mature pollen provides evidence for developmental priming. J. Proteome Res. 2013, 12, 4892–4903. [Google Scholar] [CrossRef] [PubMed]
- Frank, G.; Pressman, E.; Ophir, R.; Althan, L.; Shaked, R.; Freedman, M.; Shen, S.; Firon, N. Transcriptional profiling of maturing tomato (Solanum lycopersicum L.) microspores reveals the involvement of heat shock proteins, ROS scavengers, hormones, and sugars in the heat stress response. J. Exp. Bot. 2009, 60, 3891–3908. [Google Scholar] [CrossRef] [PubMed]
- Volkov, R.A.; Panchuk, I.I.; Schoffl, F. Small heat shock proteins are differentially regulated during pollen development and following heat stress in tobacco. Plant. Mol. Biol. 2005, 57, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.Z.; Xia, C.; Liu, X.L.; Dou, X.Y.; Wang, W.; Chen, L.Q.; Zhang, X.Q.; Xie, L.F.; He, L.; Ma, X.; et al. A mutation in Thermosensitive Male Sterile 1, encoding a heat shock protein with DnaJ and PDI domains, leads to thermosensitive gametophytic male sterility in Arabidopsis. Plant. J. 2009, 57, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Fragkostefanakis, S.; Mesihovic, A.; Hu, Y.; Schleiff, E. Unfolded protein response in pollen development and heat stress tolerance. Plant. Reprod. 2016, 29, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Doerfler, H.; Jegadeesan, S.; Ghatak, A.; Pressman, E.; Castillejo, M.A.; Wienkoop, S.; Egelhofer, V.; Firon, N.; Weckwerth, W. Heat-Treatment-Responsive Proteins in Different Developmental Stages of Tomato Pollen Detected by Targeted Mass Accuracy Precursor Alignment (tMAPA). J. Proteome Res. 2015, 14, 4463–4471. [Google Scholar] [CrossRef] [PubMed]
- Bita, C.E.; Zenoni, S.; Vriezen, W.H.; Mariani, C.; Pezzotti, M.; Gerats, T. Temperature stress differentially modulates transcription in meiotic anthers of heat-tolerant and heat-sensitive tomato plants. BMC Genomics 2011, 12, 384. [Google Scholar] [CrossRef]
- Zhang, H.; Liang, W.Q.; Yang, X.J.; Luo, X.; Jiang, N.; Ma, H.; Zhang, D.B. Carbon Starved Anther Encodes a MYB Domain Protein That Regulates Sugar Partitioning Required for Rice Pollen Development. Plant. Cell 2010, 22, 672–689. [Google Scholar] [CrossRef]
- Jin, K.; Zhong, L.; Tan, Y.P.; Wan, C.X.; Li, S.Q.; Zhu, Y.G. Different gene expression patterns of sucrose-starch metabolism during pollen maturation in cytoplasmic male-sterile and male-fertile lines of rice. Physiol. Plant 2010, 130, 136–147. [Google Scholar]
- Castro, A.J.; Clement, C. Sucrose and starch catabolism in the anther of Lilium during its development: A comparative study among the anther wall, locular fluid and microspore/pollen fractions. Planta 2007, 225, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.; Godt, D.E.; Guivarc’h, A.; Kahmann, U.; Chriqui, D.; Roitsch, T. Induction of male sterility in plants by metabolic engineering of the carbohydrate supply. Proc. Natl. Acad. Sci. USA 2001, 98, 6522–6527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, R.; Chamusco, K.C.; Chourey, P.S. Starch biosynthesis during pollen maturation is associated with altered patterns of gene expression in maize. Plant. Physiol. 2002, 130. [Google Scholar] [CrossRef] [PubMed]
- Piffanelli, P.; Ross, J.H.E.; Murphy, D.J. Biogenesis and function of the lipidic structures of pollen grains. Sex. Plant. Reprod. 1998, 11, 65–80. [Google Scholar] [CrossRef]
- Sreelakshmi, Y.; Sharma, R. Differential regulation of phenylalanine ammonia lyase activity and protein level by light in tomato seedlings. Plant. Physiol. Biochem. 2008, 46, 444–451. [Google Scholar] [CrossRef]
- Buer, C.S.; Imin, N.; Djordjevic, M.A. Flavonoids: New roles for old molecules. J. Integr. Plant. Biol. 2010, 52, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Nagel, C.; Taylor, L.P. Biochemical complementation of chalcone synthase mutants defines a role for flavonols in functional pollen. Proc. Natl. Acad. Sci. USA 1992, 89, 7213–7217. [Google Scholar] [CrossRef]
- Kishitani, S.; Yomoda, A.; Konno, N.; Tanaka, Y. Involvement of Phenylalanine Ammonia-Lyase in the Development of Pollen in Broccoli (Brassica Oleracea L.). Sex. Plant. Reprod. 1993, 6, 244–248. [Google Scholar] [CrossRef]
- Li, H.; Zhang, D. Biosynthesis of anther cuticle and pollen exine in rice. Plant. Signal. Behav. 2010, 5, 1121–1123. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.C.; Shi, J.X.; Liang, W.Q.; Xue, F.Y.; Luo, Q.; Zhu, L.; Qu, G.R.; Chen, M.J.; Schreiber, L.; Zhang, D.B. Two ATP Binding Cassette G Transporters, Rice ATP Binding Cassette G26 and ATP Binding Cassette G15, Collaboratively Regulate Rice Male Reproduction. Plant. Physiol. 2015, 169, 2064–2079. [Google Scholar]
- Nawrath, C. Unraveling the complex network of cuticular structure and function. Curr. Opin. Plant. Biol. 2006, 9, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Kurdyukov, S.; Faust, A.; Trenkamp, S.; Bar, S.; Franke, R.; Efremova, N.; Tietjen, K.; Schreiber, L.; Saedler, H.; Yephremov, A. Genetic and biochemical evidence for involvement of HOTHEAD in the biosynthesis of long-chain alpha-,omega-dicarboxylic fatty acids and formation of extracellular matrix. Planta 2006, 224, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, S.S.; Liu, Y.Q.; Ling, S.; Chen, C.S.; Yao, J.L. HOTHEAD-Like HTH1 is Involved in Anther Cutin Biosynthesis and is Required for Pollen Fertility in Rice. Plant. Cell Physiol. 2017, 58, 1238–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.J.; Tian, H.; Wang, S.Z.; Shao, J.Z.; Zheng, Y.Z.; Zhang, H.Y.; Guo, L.; Ding, Y. Pollen developmental defects in ZD-CMS rice line explored by cytological, molecular and proteomic approaches. J. Proteomics 2014, 108, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Damaris, R.N.; Li, M.; Liu, Y.L.; Chen, X.; Murage, H.; Yang, P.F. A proteomic analysis of salt stress response in seedlings of two African rice cultivars. Biochim. Biophys. Acta 2016, 1864, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Zhou, R.; Feng, Q.; Wang, Q.; Wang, J.; Liu, S. IQuant: An automated pipeline for quantitative proteomics based upon isobaric tags. Proteomics 2014, 14, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Onsongo, G.; Stone, M.D.; Van Riper, S.K.; Chilton, J.; Wu, B.; Higgins, L.; Lund, T.C.; Carlis, J.V.; Griffin, T.J. LTQ-iQuant: A freely available software pipeline for automated and accurate protein quantification of isobaric tagged peptide data from LTQ instruments. Proteomics 2010, 10, 3533–3538. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Briefings. Bioinf. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biology 2010, 11, R106. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Description | Protein Fold Change | p-Value | |
|---|---|---|---|---|
| SP2/FP2 | SP3/FP3 | |||
| LOC_Os01g24680.1 | 3-hydroxyacyl-CoA dehydrogenase, putative, expressed | − | 0.81 | 1.0 × 10 −4 |
| LOC_Os06g48180.1 | glycosyl hydrolases family 16 (GH16), putative, expressed | 0.51 | − | 8.0 × 10 −4 |
| LOC_Os05g04380.1 | peroxidase precursor, putative, expressed | 0.51 | − | 1.0 × 10 −4 |
| LOC_Os03g48770.1 | Cupin domain containing protein, expressed | 0.48 | − | 9.0 × 10 −4 |
| LOC_Os02g32980.1 | Cupin domain containing protein, expressed | 0.42 | − | 1.0 × 10 −4 |
| LOC_Os06g43640.1 | Ser/Thr protein phosphatase family protein, putative, expressed | − | 0.74 | 2.1 × 10 −3 |
| LOC_Os01g18170.1 | Cupin domain containing protein, expressed | 0.69 | − | 1.9 × 10 −2 |
| Protein ID | Description | Protein Fold Change | p-Value | |
|---|---|---|---|---|
| SP2/FP2 | SP3/FP3 | |||
| LOC_Os01g08860.1 | hsp20/alpha crystallin family protein, putative, expressed | 3.25 | - | 1.0 × 10 −4 |
| LOC_Os02g52150.2 | heat shock 22 kDa protein, mitochondrial precursor, putative, expressed | 2.19 | - | 2.0 × 10 −4 |
| LOC_Os03g60620.1 | DnaK family protein, putative, expressed | 0.7 | - | 1.0 × 10 −4 |
| LOC_Os04g01740.1 | heat shock protein, putative, expressed | 2.45 | - | 1.0 × 10 −4 |
| LOC_Os05g23740.1 | DnaK family protein, putative, expressed | 1.22 | - | 1.0 × 10 −4 |
| LOC_Os05g38530.1 | DnaK family protein, putative, expressed | 2.89 | - | 1.0 × 10 −4 |
| LOC_Os12g32986.1 | heat shock protein, putative, expressed | 1.4 | - | 1.0 × 10 −4 |
| LOC_Os03g45340.1 | hsp20/alpha crystallin family protein, putative, expressed | − | 0.74 | 2.4 × 10 −3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Jin, J.; Zhang, H.; Wang, Y.; Li, Q.; Zou, Y.; Huang, X.; Zhou, B.; Zhou, R.; Ding, Y. Comparative Analysis of Proteomics and Transcriptomics during Fertility Transition in a Two-Line Hybrid Rice Line Wuxiang S. Int. J. Mol. Sci. 2019, 20, 4542. https://doi.org/10.3390/ijms20184542
Chen H, Jin J, Zhang H, Wang Y, Li Q, Zou Y, Huang X, Zhou B, Zhou R, Ding Y. Comparative Analysis of Proteomics and Transcriptomics during Fertility Transition in a Two-Line Hybrid Rice Line Wuxiang S. International Journal of Molecular Sciences. 2019; 20(18):4542. https://doi.org/10.3390/ijms20184542
Chicago/Turabian StyleChen, Hao, Jing Jin, Hongyuan Zhang, Ying Wang, Qian Li, Yu Zou, Xingguo Huang, Baojin Zhou, Ruo Zhou, and Yi Ding. 2019. "Comparative Analysis of Proteomics and Transcriptomics during Fertility Transition in a Two-Line Hybrid Rice Line Wuxiang S" International Journal of Molecular Sciences 20, no. 18: 4542. https://doi.org/10.3390/ijms20184542
APA StyleChen, H., Jin, J., Zhang, H., Wang, Y., Li, Q., Zou, Y., Huang, X., Zhou, B., Zhou, R., & Ding, Y. (2019). Comparative Analysis of Proteomics and Transcriptomics during Fertility Transition in a Two-Line Hybrid Rice Line Wuxiang S. International Journal of Molecular Sciences, 20(18), 4542. https://doi.org/10.3390/ijms20184542