Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions

 , , ,
, , ,
Abstract
:1. Introduction
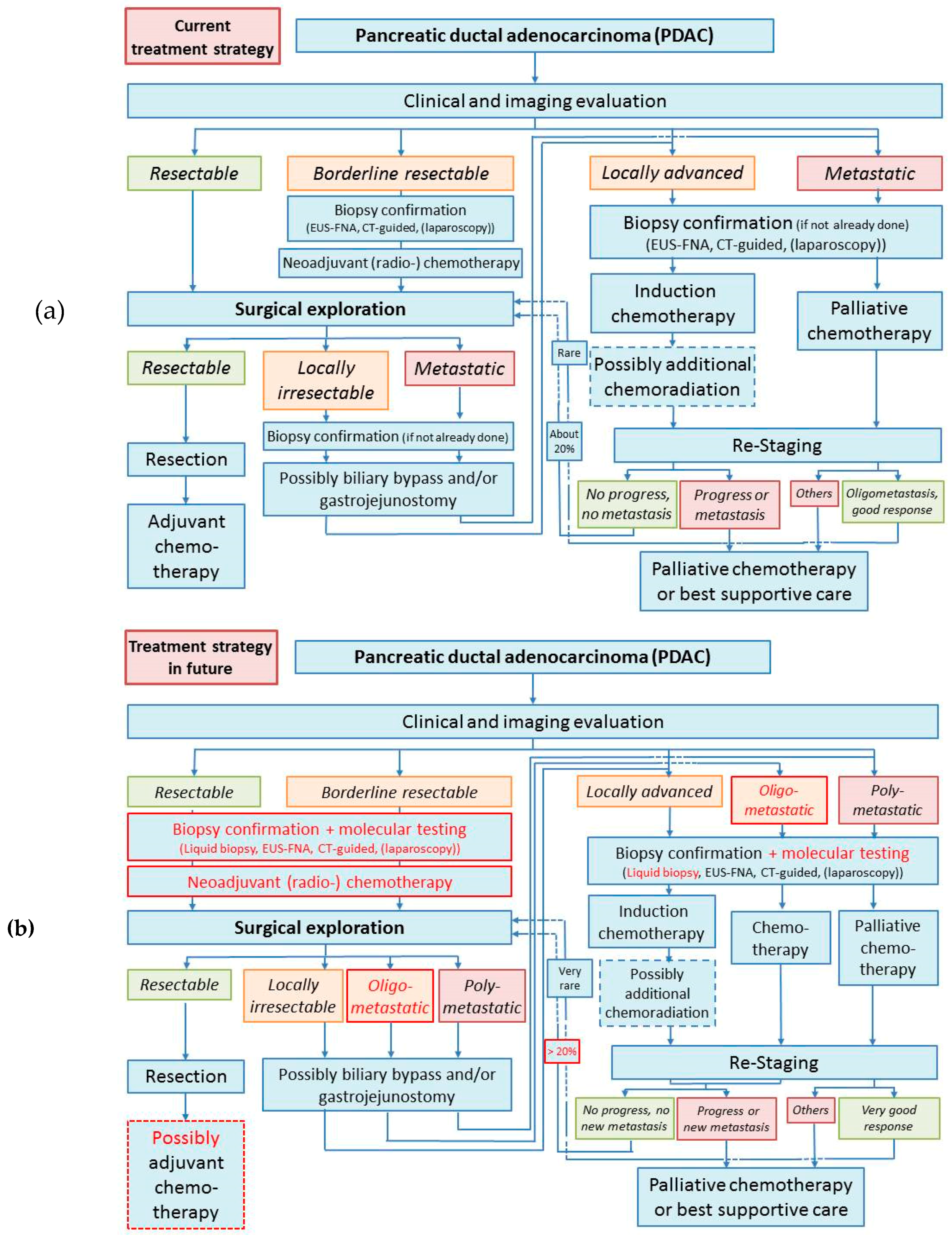
2. Current Management of Pancreatic Cancer
2.1. Management of Resectable Pancreatic Cancers
2.2. Management of Borderline Resectable Pancreatic Cancers
2.3. Management of Locally Advanced and Metastatic Pancreatic Cancer
2.4. Palliative Care
2.5. Conclusion/Future Directions
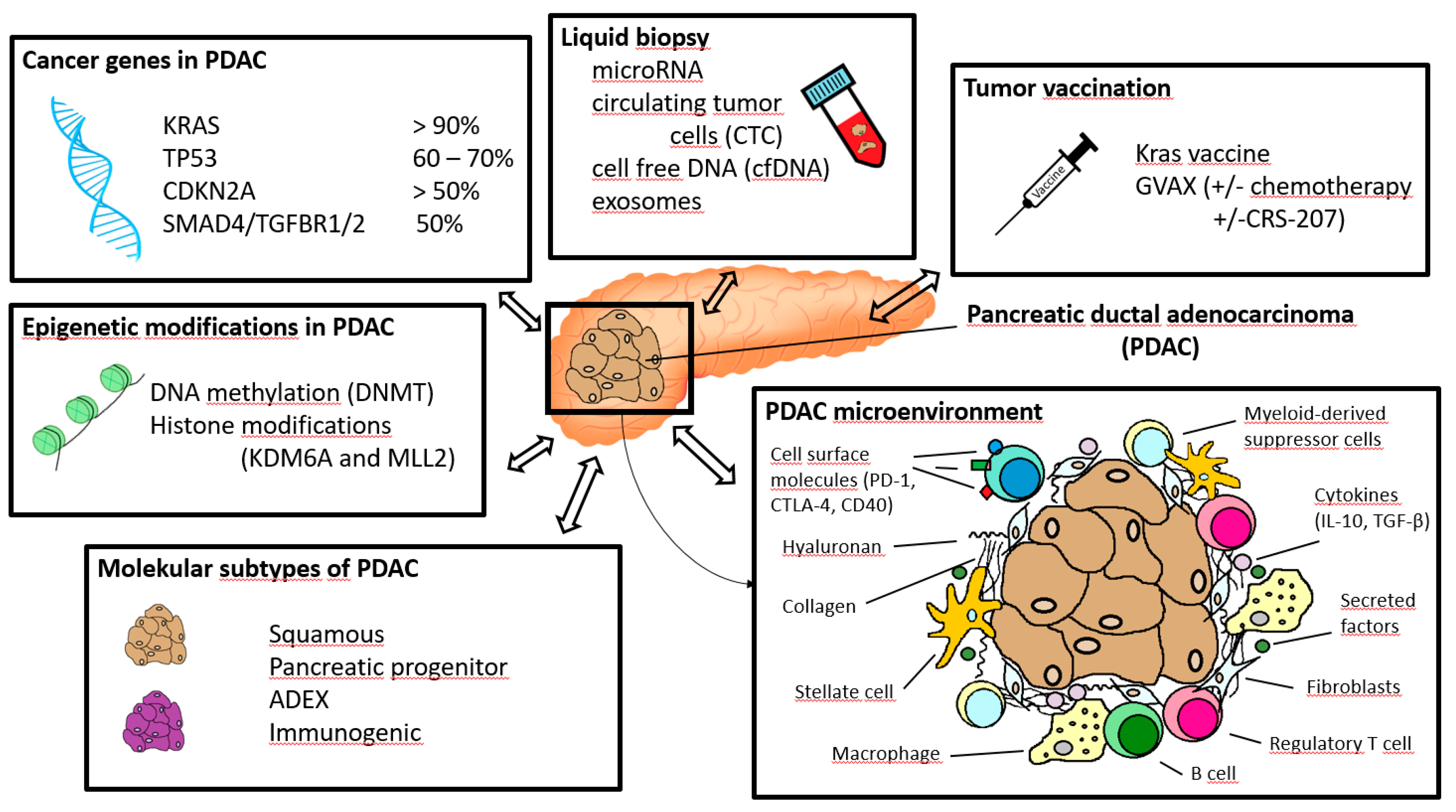
3. Immunotherapy and Other State-of-the-Art Molecular Options
3.1. Immunotherapy
3.2. Molecular Pathology of Pancreatic Ductal Adenocarcinoma
3.2.1. Molecular Subtypes of Pancreatic Cancers and Other Considerations Based on Genomic and Transcriptomic Analyses
3.2.2. Epigenetic Modifications in the Tumorigenesis of PDACs
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| EUS-FNA | Endoscopic ultrasound-guided fine needle aspiration |
| CRT | Chemoradiotherapy |
| ECOG | Eastern Co-operative Oncology Group |
| NK cells | Natural killer cells |
| APC | Antigen presenting cells |
| MHC | Major histocompatibility complex |
| PD-1 | Programed cell death-1 |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein-4 |
| FAP | Fibroblast-associated protein |
| CAF | Cancer-associated fibroblast |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| PanIN | Pancreatic intraepithelial neoplasm |
| CDKN2A | Cyclin dependent kinase inhibitor 2A |
| TGF-β | Transforming growth factor β |
| ADEX | Aberrantly differentiated endocrine exocrine |
| HMT | Histone methyltransferase |
| HDM | Histone demethylase |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| UNR | Upstream of N-ras |
| PIWI | P-element-induced wimpy testis |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Keane, M.G.; Horsfall, L.; Rait, G.; Pereira, S.P. A case—Control study comparing the incidence of early symptoms in pancreatic and biliary tract cancer. BMJ Open 2014, 4, 005720. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McCallister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, K.; Ejaz, A.; He, J. Technical aspects of pancreaticoduodenectomy and their outcomes. Chin. Clin. Oncol. 2017, 6, 901. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Klimstra, D.S.; Brennan, M.F. Long-term survival after curative resection for pancreatic ductal adenocarcinoma. Clinicopathologic analysis of 5-year survivors. Ann. Surg. 1996, 223, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.; Krug, J.; Yu, J.; Zyromski, N.; Schmidt, C.; Jacobson, L.; Madura, J.; Wiebke, E.; Lillemoe, K. A Margin-Negative R0 Resection Accomplished with Minimal Postoperative Complications Is the Surgeon’s Contribution to Long-Term Survival in Pancreatic Cancer. J. Gastrointest. Surg. 2006, 10, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Quarto, G.; Perrotta, S.; Furino, E.; Amato, B.; Bianco, T.; De Palma, G.; Forestieri, P.; Benassai, G.; Benassai, G.L.; Benassai, G. Long-term survival after curative resection for pancreatic ductal adenocarcinoma—Surgical treatment. Int. J. Surg. 2015, 21, S1–S3. [Google Scholar]
- La Torre, M.; Nigri, G.; Ferrari, L.; Cosenza, G.; Ravaioli, M.; Ramacciato, G. Hospital volume, margin status, and long-term survival after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am. Surg. 2012, 78, 225–229. [Google Scholar]
- Nimptsch, U.; Krautz, C.; Weber, G.F.; Mansky, T.; Grützmann, R. Nationwide In-hospital Mortality Following Pancreatic Surgery in Germany is Higher than Anticipated. Ann. Surg. 2016, 264, 1–1090. [Google Scholar] [CrossRef] [PubMed]
- Krautz, C.; Nimptsch, U.; Weber, G.F.; Mansky, T.; Grützmann, R. Effect of Hospital Volume on In-hospital Morbidity and Mortality Following Pancreatic Surgery in Germany. Ann. Surg. 2018, 267, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Bakkevold, K.; Arnesjø, B.; Dahl, O.; Kambestad, B. Adjuvant combination chemotherapy (AMF) following radical resection of carcinoma of the pancreas and papilla of vater—Results of a controlled, prospective, randomised multicentre study. Eur. J. Cancer 1993, 29, 698–703. [Google Scholar] [CrossRef]
- Study Group of Surgical Adjuvant Therapy for Carcinomas of the Pancreas and Biliary Tract; Takada, T.; Amano, H.; Yasuda, H.; Nimura, Y.; Matsushiro, T.; Kato, H.; Nagakawa, T.; Nakayama, T. Is postoperative adjuvant chemotherapy useful for gallbladder carcinoma? A phase III multicenter prospective randomized controlled trial in patients with resected pancreaticobiliary carcinoma. Cancer 2002, 95, 1685–1695. [Google Scholar] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H.; Beger, H.; Fernandez-Cruz, L.; Dervenis, C.; Lacaine, F.; et al. A Randomized Trial of Chemoradiotherapy and Chemotherapy after Resection of Pancreatic Cancer. N. Engl. J. Med. 2004, 350, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Kosuge, T.; Kiuchi, T.; Mukai, K.; Kakizoe, T. A Multicenter Randomized Controlled Trial to Evaluate the Effect of Adjuvant Cisplatin and 5-Fluorouracil Therapy after Curative Resection in Cases of Pancreatic Cancer. Jpn. J. Clin. Oncol. 2006, 36, 159–165. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs. observation in patients undergoing curative-intent resection of pancreatic cancer: A randomized controlled trial. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef]
- Yoshitomi, H.; Togawa, A.; Kimura, F.; Ito, H.; Shimizu, H.; Yoshidome, H.; Otsuka, M.; Kato, A.; Nozawa, S.; Furukawa, K.; et al. A randomized phase II trial of adjuvant chemotherapy with uracil/tegafur and gemcitabine versus gemcitabine alone in patients with resected pancreatic cancer. Cancer 2008, 113, 2448–2456. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Stocken, D.D.; Smith, C.T.; Bassi, C.; Ghaneh, P.; Owen, E.; Moore, M.; Padbury, R.; Doi, R.; Smith, D.; et al. Adjuvant 5-fluorouracil and folinic acid vs observation for pancreatic cancer: Composite data from the ESPAC-1 and -3(v1) trials. Br. J. Cancer 2009, 100, 246–250. [Google Scholar] [CrossRef]
- Ueno, H.; Kosuge, T.; Matsuyama, Y.; Yamamoto, J.; Nakao, A.; Egawa, S.; Doi, R.; Monden, M.; Hatori, T.; Tanaka, M.; et al. A randomised phase III trial comparing gemcitabine with surgery-only in patients with resected pancreatic cancer: Japanese Study Group of Adjuvant Therapy for Pancreatic Cancer. Br. J. Cancer 2009, 101, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs. gemcitabine following pancreatic cancer resection: A randomized controlled trial. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Regine, W.F.; Winter, K.A.; Abrams, R.; Safran, H.; Hoffman, J.P.; Konski, A.; Benson, A.B.; Macdonald, J.S.; Rich, T.A.; Willett, C.G. Fluorouracil-based chemoradiation with either gemcitabine or fluorouracil chemotherapy after resection of pancreatic adenocarcinoma: 5-year analysis of the U.S. Intergroup/RTOG 9704 phase III trial. Ann. Surg. Oncol. 2011, 18, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Regine, W.F.; Winter, K.A.; Abrams, R.A.; Safran, H.; Hoffman, J.P.; Konski, A.; Benson, A.B.; Macdonald, J.S.; Kudrimoti, M.R.; Fromm, M.L.; et al. Fluorouracil vs. gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: A randomized controlled trial. JAMA 2008, 299, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Balzano, G.; Aprile, G.; Cereda, S.; Passoni, P.; Zerbi, A.; Tronconi, M.C.; Milandri, C.; Saletti, P.; Rognone, A.; et al. Adjuvant PEFG (Cisplatin, Epirubicin, 5-Fluorouracil, Gemcitabine) or Gemcitabine Followed by Chemoradiation in Pancreatic Cancer: A Randomized Phase II Trial. Ann. Surg. Oncol. 2012, 19, 2256–2263. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Kubota, K.; Shimizu, T.; Katoh, M. Randomized clinical trial of adjuvant chemotherapy with S-1 versus gemcitabine after pancreatic cancer resection. Br. J. Surg. 2015, 102, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Uesaka, K.; Boku, N.; Fukutomi, A.; Okamura, Y.; Konishi, M.; Matsumoto, I.; Kaneoka, Y.; Shimizu, Y.; Nakamori, S.; Sakamoto, H.; et al. Adjuvant chemotherapy of S-1 versus gemcitabine for resected pancreatic cancer: A phase 3, open-label, randomised, non-inferiority trial (JASPAC 01). Lancet 2016, 388, 248–257. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy with Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients After R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337. [Google Scholar] [CrossRef]
- Reni, M.; Balzano, G.; Zanon, S.; Zerbi, A.; Rimassa, L.; Castoldi, R.; Pinelli, D.; Mosconi, S.; Doglioni, C.; Chiaravalli, M.; et al. Safety and efficacy of preoperative or postoperative chemotherapy for resectable pancreatic adenocarcinoma (PACT-15): A randomised, open-label, phase 2-3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 413–423. [Google Scholar] [CrossRef]
- Kooby, D.A.; Gillespie, T.W.; Liu, Y.; Byrd-Sellers, J.; Landry, J.; Bian, J.; Lipscomb, J. Impact of Adjuvant Radiotherapy on Survival after Pancreatic Cancer Resection: An Appraisal of Data from the National Cancer Data Base. Ann. Surg. Oncol. 2013, 20, 3634–3642. [Google Scholar] [CrossRef] [PubMed]
- Morganti, A.G.; Falconi, M.; Van Stiphout, R.G.; Mattiucci, G.-C.; Alfieri, S.; Calvo, F.A.; Dubois, J.-B.; Fastner, G.; Herman, J.M.; Maidment, B.W.; et al. Multi-institutional Pooled Analysis on Adjuvant Chemoradiation in Pancreatic Cancer. Int. J. Radiat. Oncol. 2014, 90, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Rutter, C.E.; Park, H.S.; Corso, C.D.; Lester-Coll, N.H.; Mancini, B.R.; Yeboa, D.N.; Johung, K.L. Addition of radiotherapy to adjuvant chemotherapy is associated with improved overall survival in resected pancreatic adenocarcinoma: An analysis of the National Cancer Data Base. Cancer 2015, 121, 4141–4149. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, A.; Kunieda, E. Effect of adjuvant radiotherapy on survival in resected pancreatic cancer: A propensity score surveillance, epidemiology, and end results database analysis. J. Surg. Oncol. 2014, 110, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-C.; Chien, K.-L.; Lin, Y.-L.; Wu, M.-S.; Lin, J.-T.; Wang, H.-P.; Tu, Y.-K. Adjuvant treatments for resected pancreatic adenocarcinoma: A systematic review and network meta-analysis. Lancet Oncol. 2013, 14, 1095–1103. [Google Scholar] [CrossRef]
- Palmer, D.H.; Stocken, D.D.; Hewitt, H.; Markham, C.E.; Hassan, A.B.; Johnson, P.J.; Buckels, J.A.C.; Bramhall, S.R. A Randomized Phase 2 Trial of Neoadjuvant Chemotherapy in Resectable Pancreatic Cancer: Gemcitabine Alone Versus Gemcitabine Combined with Cisplatin. Ann. Surg. Oncol. 2007, 14, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Sahora, K.; Schindl, M.; Kuehrer, I.; Eisenhut, A.; Werba, G.; Brostjan, C.; Telek, B.; Ba’Ssalamah, A.; Stift, J.; Schoppmann, S.F.; et al. A phase II trial of two durations of Bevacizumab added to neoadjuvant gemcitabine for borderline and locally advanced pancreatic cancer. Anticancer Res. 2014, 34, 2377–2384. [Google Scholar]
- Landry, J.; Catalano, P.J.; Staley, C.; Harris, W.; Hoffman, J.; Talamonti, M.; Xu, N.; Cooper, H.; Benson, A.B., III. Randomized phase II study of gemcitabine plus radiotherapy versus gemcitabine, 5-fluorouracil, and cisplatin followed by radiotherapy and 5-fluorouracil for patients with locally advanced, potentially resectable pancreatic adenocarcinoma. J. Surg. Oncol. 2010, 101, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golcher, H.; Brunner, T.B.; Witzigmann, H.; Marti, L.; Bechstein, W.O.; Bruns, C.; Jungnickel, H.; Schreiber, S.; Grabenbauer, G.G.; Meyer, T.; et al. Neoadjuvant chemoradiation therapy with gemcitabine/cisplatin and surgery versus immediate surgery in resectable pancreatic cancer. Strahlenther. Onkol. 2015, 191, 7–16. [Google Scholar] [CrossRef]
- Casadei, R.; Di Marco, M.; Ricci, C.; Santini, D.; Serra, C.; Calculli, L.; D’Ambra, M.; Guido, A.; Morselli-Labate, A.M.; Minni, F. Neoadjuvant Chemoradiotherapy and Surgery Versus Surgery Alone in Resectable Pancreatic Cancer: A Single-Center Prospective, Randomized, Controlled Trial Which Failed to Achieve Accrual Targets. J. Gastrointest. Surg. 2015, 19, 1802–1812. [Google Scholar] [CrossRef]
- Tzeng, C.-W.D.; Fleming, J.B.; Lee, J.E.; Wang, X.; Pisters, P.W.T.; Vauthey, J.-N.; Varadhachary, G.; Wolff, R.A.; Katz, M.H.G. Yield of clinical and radiographic surveillance in patients with resected pancreatic adenocarcinoma following multimodal therapy. HPB 2012, 14, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, C.-W.D.; Abbott, D.E.; Cantor, S.B.; Fleming, J.B.; Lee, J.E.; Pisters, P.W.T.; Varadhachary, G.R.; Abbruzzese, J.L.; Wolff, R.A.; Ahmad, S.A.; et al. Frequency and Intensity of Postoperative Surveillance After Curative Treatment of Pancreatic Cancer: A Cost-Effectiveness Analysis. Ann. Surg. Oncol. 2013, 20, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Schorn, S.; Demir, I.E.; Reyes, C.M.; Saricaoglu, C.; Samm, N.; Schirren, R.; Tieftrunk, E.; Hartmann, D.; Friess, H.; Ceyhan, G.O. The impact of neoadjuvant therapy on the histopathological features of pancreatic ductal adenocarcinoma—A systematic review and meta-analysis. Cancer Treat. Rev. 2017, 55, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lutfi, W.; Talamonti, M.S.; Kantor, O.; Wang, C.-H.; Liederbach, E.; Stocker, S.J.; Bentrem, D.J.; Roggin, K.K.; Winchester, D.J.; Marsh, R.; et al. Perioperative chemotherapy is associated with a survival advantage in early stage adenocarcinoma of the pancreatic head. Surgery 2016, 160, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Shubert, C.R.; Bergquist, J.R.; Groeschl, R.T.; Habermann, E.B.; Wilson, P.M.; Truty, M.J.; Smoot, R.L.; Kendrick, M.L.; Nagorney, D.M.; Farnell, M.B. Overall survival is increased among stage III pancreatic adenocarcinoma patients receiving neoadjuvant chemotherapy compared to surgery first and adjuvant chemotherapy: An intention to treat analysis of the National Cancer Database. Surgery 2016, 160, 1080–1096. [Google Scholar] [CrossRef]
- Verma, V.; Li, J.; Lin, C. Neoadjuvant Therapy for Pancreatic Cancer: Systematic Review of Postoperative Morbidity, Mortality, and Complications. Am. J. Clin. Oncol. 2016, 39, 302–313. [Google Scholar] [CrossRef]
- Burris, H.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Christine Cripps, M.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Huguier, M.; Barrier, A.; Valinas, R.; Flahault, A.; Adloff, M.; Pezet, D.; Jaeck, D.; Millat, B. Randomized trial of 5-fluorouracil, leucovorin and cisplatin in advanced pancreatic cancer. Hepatogastroenterology 2001, 48, 875–878. [Google Scholar]
- Ducreux, M.; Rougier, P.; Pignon, J.-P.; Douillard, J.-Y.; Seitz, J.-F.; Bugat, R.; Bosset, J.-F.; Merouche, Y.; Raoul, J.-L.; Ychou, M.; et al. A randomised trial comparing 5-FU with 5-FU plus cisplatin in advanced pancreatic carcinoma. Ann. Oncol. 2002, 13, 1185–1191. [Google Scholar] [CrossRef]
- Colucci, G.; Giuliani, F.; Gebbia, V.; Biglietto, M.; Rabitti, P.; Uomo, G.; Cigolari, S.; Testa, A.; Maiello, E.; Lopez, M. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: A prospective, randomized phase III study of the Gruppo Oncologia dell’Italia Meridionale. Cancer 2002, 94, 902–910. [Google Scholar] [CrossRef]
- Scheithauer, W.; Schüll, B.; Ulrich-Pur, H.; Raderer, M.; Haider, K.; Depisch, D.; Kornek, G.V.; Schmid, K.; Kwasny, W.; Schneeweiss, B.; et al. Biweekly high-dose gemcitabine alone or in combination with capecitabine in patients with metastatic pancreatic adenocarcinoma: A randomized phase II trial. Ann. Oncol. 2003, 14, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.; Plunkett, W.; Van Haperen, V.R.; Hainsworth, J.; Hochster, H.; Lenzi, R.; Abbruzzese, J. Randomized Phase II Comparison of Dose-Intense Gemcitabine: Thirty-Minute Infusion and Fixed Dose Rate Infusion in Patients with Pancreatic Adenocarcinoma. J. Clin. Oncol. 2003, 21, 3402–3408. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Mitry, E.; Ould-Kaci, M.; Boige, V.; Seitz, J.F.; Bugat, R.; Breau, J.L.; Bouché, O.; Etienne, P.L.; Tigaud, J.M.; et al. Randomized phase II study evaluating oxaliplatin alone, oxaliplatin combined with infusional 5-FU, and infusional 5-FU alone in advanced pancreatic carcinoma patients. Ann. Oncol. 2004, 15, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Louvet, C. Gemcitabine in Combination with Oxaliplatin Compared with Gemcitabine Alone in Locally Advanced or Metastatic Pancreatic Cancer: Results of a GERCOR and GISCAD Phase III Trial. J. Clin. Oncol. 2005, 23, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Quietzsch, D.; Gieseler, F.; Gonnermann, M.; Schönekäs, H.; Rost, A.; Neuhaus, H.; Haag, C.; Clemens, M.; Heinrich, B.; et al. Randomized Phase III Trial of Gemcitabine Plus Cisplatin Compared with Gemcitabine Alone in Advanced Pancreatic Cancer. J. Clin. Oncol. 2006, 24, 3946–3952. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared with Gemcitabine Alone in Patients with Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, R.; Bodoky, G.; Ruhstaller, T.; Glimelius, B.; Bajetta, E.; Schüller, J.; Saletti, P.; Bauer, J.; Figer, A.; Pestalozzi, B.; et al. Gemcitabine Plus Capecitabine Compared with Gemcitabine Alone in Advanced Pancreatic Cancer: A Randomized, Multicenter, Phase III Trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J. Clin. Oncol. 2007, 25, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Boeck, S.; Hoehler, T.; Seipelt, G.; Mahlberg, R.; Wein, A.; Hochhaus, A.; Boeck, H.-P.; Schmid, B.; Kettner, E.; Stauch, M.; et al. Capecitabine plus oxaliplatin (CapOx) versus capecitabine plus gemcitabine (CapGem) versus gemcitabine plus oxaliplatin (mGemOx): Final results of a multicenter randomized phase II trial in advanced pancreatic cancer. Ann. Oncol. 2008, 19, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III Randomized Comparison of Gemcitabine Versus Gemcitabine Plus Capecitabine in Patients with Advanced Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef]
- Poplin, E.; Feng, Y.; Berlin, J.; Rothenberg, M.L.; Hochster, H.; Mitchell, E.; Alberts, S.; O’Dwyer, P.; Haller, D.; Catalano, P.; et al. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-min infusion) in patients with pancreatic carcinoma E6201: A trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2009, 27, 3778–3785. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Tempero, M.A.; Niedzwiecki, D.; Hollis, D.R.; Kindler, H.L.; Cusnir, M.; Enzinger, P.C.; Gorsch, S.M.; Goldberg, R.M.; Mayer, R.J. Randomized Phase II Study of Gemcitabine Administered at a Fixed Dose Rate or in Combination with Cisplatin, Docetaxel, or Irinotecan in Patients with Metastatic Pancreatic Cancer: CALGB 89904. J. Clin. Oncol. 2009, 27, 5506–5512. [Google Scholar] [CrossRef] [PubMed]
- Colucci, G.; Labianca, R.; Di Costanzo, F.; Gebbia, V.; Cartenì, G.; Massidda, B.; Dapretto, E.; Manzione, L.; Piazza, E.; Sannicolo, M.; et al. Randomized Phase III Trial of Gemcitabine Plus Cisplatin Compared with Single-Agent Gemcitabine as First-Line Treatment of Patients with Advanced Pancreatic Cancer: The GIP-1 Study. J. Clin. Oncol. 2010, 28, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Dahan, L.; Bonnetain, F.; Ychou, M.; Mitry, E.; Gasmi, M.; Raoul, J.-L.; Cattan, S.; Phelip, J.-M.; Hammel, P.; Chauffert, B.; et al. Combination 5-fluorouracil, folinic acid and cisplatin (LV5FU2-CDDP) followed by gemcitabine or the reverse sequence in metastatic pancreatic cancer: Final results of a randomised strategic phase III trial (FFCD 0301). Gut 2010, 59, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardiere, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaka, M.; Matsumura, Y.; Ishii, H.; Omuro, Y.; Itoi, T.; Mouri, H.; Hanada, K.; Kimura, Y.; Maetani, I.; Okabe, Y.; et al. Randomized phase II study of gemcitabine and S-1 combination versus gemcitabine alone in the treatment of unresectable advanced pancreatic cancer (Japan Clinical Cancer Research Organization PC-01 study). Cancer Chemother. Pharmacol. 2012, 69, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Sasaki, T.; Sasahira, N.; Tsujino, T.; Toda, N.; Kogure, H.; Matsubara, S.; Ito, Y.; Togawa, O.; et al. A multicentre randomised phase II trial of gemcitabine alone vs gemcitabine and S-1 combination therapy in advanced pancreatic cancer: GEMSAP study. Br. J. Cancer 2012, 106, 1934–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.; Wu, C.-Y.; Wang, J.P.; Lee, R.-C.; Lee, W.-P.; Li, C.-P. A randomized controlled trial of gemcitabine plus cisplatin versus gemcitabine alone in the treatment of metastatic pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 72, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, H.; Ioka, T.; Ikeda, M.; Ohkawa, S.; Yanagimoto, H.; Boku, N.; Fukutomi, A.; Sugimori, K.; Baba, H.; Yamao, K.; et al. Randomized Phase III Study of Gemcitabine Plus S-1, S-1 Alone, or Gemcitabine Alone in Patients with Locally Advanced and Metastatic Pancreatic Cancer in Japan and Taiwan: GEST Study. J. Clin. Oncol. 2013, 31, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Okusaka, T.; Miyakawa, H.; Fujii, H.; Nakamori, S.; Satoh, T.; Hamamoto, Y.; Ito, T.; Maguchi, H.; Matsumoto, S.; Ueno, H.; et al. Updated results from GEST study: A randomized, three-arm phase III study for advanced pancreatic cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Ishihara, T.; Hirata, N.; Ozawa, F.; Ohshima, T.; Azemoto, R.; Shimura, K.; Nihei, T.; Nishino, T.; Nakagawa, A.; et al. Randomized controlled study of gemcitabine plus S-1 combination chemotherapy versus gemcitabine for unresectable pancreatic cancer. Cancer Chemother. Pharmacol. 2014, 73, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Petrioli, R.; Roviello, G.; Fiaschi, A.I.; Laera, L.; Marrelli, D.; Roviello, F.; Francini, E. Gemcitabine, oxaliplatin, and capecitabine (GEMOXEL) compared with gemcitabine alone in metastatic pancreatic cancer: A randomized phase II study. Cancer Chemother. Pharmacol. 2015, 75, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Wu, C.-Y.; Yeh, Y.-C.; Shyr, Y.-M.; Wu, Y.-Y.; Kuo, C.-Y.; Hung, Y.-P.; Chen, M.-H.; Lee, W.-P.; Luo, J.-C.; et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: A randomized, open-label, prospective trial. Oncotarget 2015, 6, 18162–18173. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Chung, M.J.; Park, J.Y.; Bang, S.; Park, S.W.; Kim, H.G.; Noh, M.H.; Lee, S.H.; Kim, Y.-T.; Kim, H.J.; et al. A randomized, multicenter, phase III study of gemcitabine combined with capecitabine versus gemcitabine alone as first-line chemotherapy for advanced pancreatic cancer in South Korea. Medicine 2017, 96, e5702. [Google Scholar] [CrossRef] [PubMed]
- Nagrial, A.; Sjoquist, K.; O’Connor, C.A.; Chantrill, L.; Biankin, A.V.; Scholten, R.J.; Yip, D.; Chin, V. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database Syst. Rev. 2018, 2018, 3. [Google Scholar]
- Huguet, F.; Girard, N.; Guerche, C.S.-E.; Hennequin, C.; Mornex, F.; Azria, D.; Guerche, C.S.-E. Chemoradiotherapy in the Management of Locally Advanced Pancreatic Carcinoma: A Qualitative Systematic Review. J. Clin. Oncol. 2009, 27, 2269–2277. [Google Scholar] [CrossRef]
- Chauffert, B.; Mornex, F.; Bonnetain, F.; Rougier, P.; Mariette, C.; Bouche, O.; Bosset, J.F.; Aparicio, T.; Mineur, L.; Azzedine, A.; et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann. Oncol. 2008, 19, 1592–1599. [Google Scholar]
- Loehrer, P.J., Sr.; Feng, Y.; Cardenes, H.; Wagner, L.; Brell, J.M.; Cella, D.; Flynn, P.; Ramanathan, R.K.; Crane, C.H.; Alberts, S.R.; et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: An Eastern Cooperative Oncology Group trial. J. Clin. Oncol. 2011, 29, 4105–4112. [Google Scholar] [CrossRef]
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouché, O.; Shannon, J.; André, T.; et al. Effect of Chemoradiotherapy vs. Chemotherapy on Survival in Patients with Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine with or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef]
- Li, C.-P.; Chao, Y.; Chi, K.-H.; Chan, W.-K.; Teng, H.-C.; Lee, R.-C.; Chang, F.-Y.; Lee, S.-D.; Yen, S.-H. Concurrent chemoradiotherapy treatment of locally advanced pancreatic cancer: Gemcitabine versus 5-fluorouracil, a randomized controlled study. Int. J. Radiat. Oncol. 2003, 57, 98–104. [Google Scholar] [CrossRef]
- Wilkowski, R.; Boeck, S.; Ostermaier, S.; Sauer, R.; Herbst, M.; Fietkau, R.; Flentje, M.; Miethe, S.; Boettcher, H.D.; Scholten, T.; et al. Chemoradiotherapy with concurrent gemcitabine and cisplatin with or without sequential chemotherapy with gemcitabine/cisplatin vs chemoradiotherapy with concurrent 5-fluorouracil in patients with locally advanced pancreatic cancer—A multi-centre randomised phase II study. Br. J. Cancer 2009, 101, 1853–1859. [Google Scholar] [PubMed]
- Mukherjee, S.; Hurt, C.N.; Bridgewater, J.; Falk, S.; Cummins, S.; Wasan, H.; Crosby, T.; Jephcott, C.; Roy, R.; Radhakrishna, G.; et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): A multicentre, randomised, phase 2 trial. Lancet Oncol. 2013, 14, 317–326. [Google Scholar] [CrossRef]
- Hurt, C.N.; Falk, S.; Crosby, T.; McDonald, A.; Ray, R.; Joseph, G.; Staffurth, J.; Abrams, R.A.; Griffiths, G.; Maughan, T.; et al. Long-term results and recurrence patterns from SCALOP: A phase II randomised trial of gemcitabine- or capecitabine-based chemoradiation for locally advanced pancreatic cancer. Br. J. Cancer 2017, 116, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Rana, V.; Janjan, N.A.; Varadhachary, G.R.; Abbruzzese, J.L.; Das, P.; Delclos, M.E.; Gould, M.S.; Evans, D.B.; Wolff, R.A.; et al. Induction chemotherapy selects patients with locally advanced, unresectable pancreatic cancer for optimal benefit from consolidative chemoradiation therapy. Cancer 2007, 110, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Huguet, F.; André, T.; Hammel, P.; Artru, P.; Balosso, J.; Selle, F.; Deniaud-Alexandre, E.; Ruszniewski, P.; Touboul, E.; Labianca, R.; et al. Impact of Chemoradiotherapy After Disease Control with Chemotherapy in Locally Advanced Pancreatic Adenocarcinoma in GERCOR Phase II and III Studies. J. Clin. Oncol. 2007, 25, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Chiu, Y.-F.; Yu, J.-C.; Chen, L.-T.; Ch’Ang, H.-J. The Role of Consolidation Chemoradiotherapy in Locally Advanced Pancreatic Cancer Receiving Chemotherapy: An Updated Systematic Review and Meta-Analysis. Cancer Res. Treat. 2018, 50, 562–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasuga, A.; Hamamoto, Y.; Takeuchi, A.; Kawasaki, K.; Suzuki, T.; Hirata, K.; Sukawa, Y.; Takaishi, H.; Kanai, T. Positive relationship between subsequent chemotherapy and overall survival in pancreatic cancer: Meta-analysis of postprogression survival for first-line chemotherapy. Cancer Chemother. Pharmacol. 2017, 79, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Duffy, A.; Liewehr, D.J.; Steinberg, S.M.; Greten, T.F. Second-line treatment in advanced pancreatic cancer: A comprehensive analysis of published clinical trials. Ann. Oncol. 2013, 24, 1972–1979. [Google Scholar] [CrossRef]
- Moss, A.C.; Morris, E.; Mac Mathuna, P. Palliative biliary stents for obstructing pancreatic carcinoma. Cochrane Database Syst. Rev. 2006, 169, 500. [Google Scholar]
- Söderlund, C.; Linder, S. Covered metal versus plastic stents for malignant common bile duct stenosis: A prospective, randomized, controlled trial. Gastrointest. Endosc. 2006, 63, 986–995. [Google Scholar] [CrossRef]
- Glazer, E.S.; Hornbrook, M.C.; Krouse, R.S. A meta-analysis of randomized trials: Immediate stent placement vs. surgical bypass in the palliative management of malignant biliary obstruction. J. Pain Symptom Manag. 2014, 47, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Jeurnink, S.M.; Van Eijck, C.H.; Steyerberg, E.W.; Kuipers, E.J.; Siersema, P.D. Stent versus gastrojejunostomy for the palliation of gastric outlet obstruction: A systematic review. BMC Gastroenterol. 2007, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Gurusamy, K.S.; Kumar, S.; Davidson, B.R. Prophylactic gastrojejunostomy for unresectable periampullary carcinoma. Cochrane Database Syst. Rev. 2013, 6, CD008533. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Yu, Z.; Zeng, J.X.; Lin, Y.; Yu, T.; Min, X.H.; Yuan, Y.H.; Chen, Q.K. Celiac plexus block for treatment of pain associated with pancreatic cancer: A meta-analysis. Pain Pract. 2014, 14, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Buwenge, M.; Macchia, G.; Arcelli, A.; Frakulli, R.; Fuccio, L.; Guerri, S.; Grassi, E.; Cammelli, S.; Cellini, F.; Morganti, A.G. Stereotactic radiotherapy of pancreatic cancer: A systematic review on pain relief. J. Pain Res. 2018, 11, 2169–2178. [Google Scholar] [CrossRef]
- Gilliland, T.M.; Villafane-Ferriol, N.; Shah, K.P.; Shah, R.M.; Cao, H.S.T.; Massarweh, N.N.; Silberfein, E.J.; Choi, E.A.; Hsu, C.; McELHANY, A.L.; et al. Nutritional and Metabolic Derangements in Pancreatic Cancer and Pancreatic Resection. Nutrients 2017, 9, 243. [Google Scholar] [CrossRef]
- Lee, A.Y.; Levine, M.N.; Baker, R.I.; Kakkar, A.K.; Prins, M.; Rickles, F.R.; Julian, J.A.; Haley, S.; Kovacs, M.J.; Bowden, C.; et al. Low-Molecular-Weight Heparin versus a Coumarin for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer. N. Engl. J. Med. 2003, 349, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, U.; Opitz, B.; Deutschinoff, G.; Stauch, M.; Reitzig, P.C.; Hahnfeld, S.; Müller, L.; Grunewald, M.; Stieler, J.M.; Sinn, M.; et al. Efficacy of Prophylactic Low–Molecular Weight Heparin for Ambulatory Patients with Advanced Pancreatic Cancer: Outcomes from the CONKO-004 Trial. J. Clin. Oncol. 2015, 33, 2028–2034. [Google Scholar] [CrossRef]
- Wormann, S.M.; Diakopoulos, K.N.; Lesina, M.; Algul, H. The immune network in pancreatic cancer development and progression. Oncogene 2014, 33, 2956–2967. [Google Scholar] [CrossRef]
- Sideras, K.; Braat, H.; Kwekkeboom, J.; Van Eijck, C.; Peppelenbosch, M.; Sleijfer, S.; Bruno, M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat. Rev. 2014, 40, 513–522. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Miyamoto, M.; Ishikawa, K.; Yoshioka, T.; Mishra, R.; Ichinokawa, K.; Matsumura, Y.; Itoh, T.; Shinohara, T.; Hirano, S.; et al. The CD40-CD154 interaction would correlate with proliferation and immune escape in pancreatic ductal adenocarcinoma. J. Surg. Oncol. 2011, 103, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [PubMed]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Alvarez, R.; Musteanu, M.; Garcia-Garcia, E.; Lopez-Casas, P.P.; Megias, D.; Guerra, C.; Muñoz, M.; Quijano, Y.; Cubillo, A.; Rodriguez-Pascual, J.; et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 2013, 109, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Wedén, S.; Klemp, M.; Gladhaug, I.P.; Møller, M.; Eriksen, J.A.; Gaudernack, G.; Buanes, T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int. J. Cancer 2011, 128, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Chapman, P.B.; Feilchenfeldt, J.; Brennan, M.F.; Capanu, M.; Gansukh, B.; Jacobs, G.; Levin, A.; Neville, D.; Kelsen, D.P.; et al. Targeting Mutated K-ras in Pancreatic Adenocarcinoma Using an Adjuvant Vaccine. Am. J. Clin. Oncol. 2011, 34, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel Allogeneic Granulocyte-Macrophage Colony-Stimulating Factor–Secreting Tumor Vaccine for Pancreatic Cancer: A Phase I Trial of Safety and Immune Activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A Lethally Irradiated Allogeneic Granulocyte-Macrophage Colony Stimulating Factor-Secreting Tumor Vaccine for Pancreatic Adenocarcinoma: A Phase II Trial of Safety, Efficacy, and Immune Activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [PubMed]
- Laheru, D.; Lutz, E.; Burke, J.; Biedrzycki, B.; Solt, S.; Onners, B.; Tartakovsky, I.; Nemunaitis, J.; Le, D.; Sugar, E.; et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: A pilot study of safety, feasibility, and immune activation. Clin. Cancer Res. 2008, 14, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin. Cancer Res. 2012, 18, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival with GVAX Pancreas Prime and Listeria Monocytogenes–Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of Ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy Converts Non-immunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Wood, L.D.; Hruban, R.H. Pathology and Molecular Genetics of Pancreatic Neoplasms. Cancer J. 2012, 18, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zhang, H.; Coloff, J.L.; Yan, H.; Wang, W.; Chen, S.; Viale, A.; Zheng, H.; Paik, J.-H.; Lim, C.; et al. Abstract A101: Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Tumor Biol. 2012, 72, A101. [Google Scholar]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.A.; Brisset, J.-C.; Zhang, Y.; Bednar, F.; Pierre, J.; Heist, K.A.; Galbán, C.J.; Galbán, S.; Di Magliano, M.P. Metastatic Pancreatic Cancer Is Dependent on Oncogenic Kras in Mice. PLoS ONE 2012, 7, e49707. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically Detected Expression of 3 Major Genes (CDKN2A/p16, TP53, and SMAD4/DPC4) Strongly Predicts Survival in Patients with Resectable Pancreatic Cancer. Ann. Surg. 2013, 258, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Caldas, C.; Hahn, S.A.; Da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 1994, 8, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar]
- Maitra, A.; Adsay, N.V.; Argani, P.; Iacobuzio-Donahue, C.; De Marzo, A.; Cameron, J.L.; Yeo, C.J.; Hruban, R.H. Multicomponent Analysis of the Pancreatic Adenocarcinoma Progression Model Using a Pancreatic Intraepithelial Neoplasia Tissue Microarray. Mod. Pathol. 2003, 16, 902–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, W.; Chianchiano, P.; Griffin, J.F.; Pittman, M.E.; Brosens, L.A.A.; Noë, M.; Yu, J.; Shindo, K.; Suenaga, M.; Rezaee, N.; et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J. Pathol. 2017, 242, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Capelli, P.; Mukai, K.; Zamboni, G.; Oda, T.; Iacono, C.; Hirohashi, S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am. J. Pathol. 1993, 142, 1534–1543. [Google Scholar] [PubMed]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-beta/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Cheng, K.-H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genome Res. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef]
- Zhou, H.; Telonis, A.G.; Jing, Y.; Xia, N.L.; Biederman, L.; Jimbo, M.; Blanco, F.; Londin, E.; Brody, J.R.; Rigoutsos, I. GPRC5A is a potential oncogene in pancreatic ductal adenocarcinoma cells that is upregulated by gemcitabine with help from HuR. Cell Death Dis. 2016, 7, e2294. [Google Scholar] [CrossRef]
- Liu, B.; Yang, H.; Pilarsky, C.; Weber, G.F. The Effect of GPRC5a on the Proliferation, Migration Ability, Chemotherapy Resistance, and Phosphorylation of GSK-3beta in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 1870. [Google Scholar] [CrossRef]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao, L.N.N.; Alqunaibit, D.; Alothman, S.; Daley, D.; Hundeyin, M.; Mani, V.R.; Barilla, R.; et al. Abstract B20: The necrosome promotes pancreatic Oncogenesis via CXCL1 and mincle-induced immune Suppression. Mol. Driv. Pancreat. Cancer Biol. Metastasis 2016, 76, B20. [Google Scholar]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 79–86. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Network CGAR. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.K.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Murphy, S.J.; Hart, S.N.; Lima, J.F.; Kipp, B.R.; Klebig, M.; Winters, J.L.; Szabo, C.; Zhang, L.; Eckloff, B.W.; Petersen, G.M.; et al. Genetic Alterations Associated with Progression from Pancreatic Intraepithelial Neoplasia to Invasive Pancreatic Tumor. Gastroenterology 2013, 145, 1098–1109.e1. [Google Scholar] [CrossRef]
- Silverman, B.R.; Shi, J. Alterations of Epigenetic Regulators in Pancreatic Cancer and Their Clinical Implications. Int. J. Mol. Sci. 2016, 17, 2138. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Song, S.; Patch, A.-M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P.; et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int. J. Cancer 2014, 135, 1110–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wang, L.; Xu, J.; Zheng, J.; Man, X.; Wu, H.; Jin, J.; Wang, K.; Xiao, H.; Li, S.; et al. Aberrant DNA methyltransferase expression in pancreatic ductal adenocarcinoma development and progression. J. Exp. Clin. Cancer Res. 2013, 32, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Zhu, Y.; Zhu, Y.; Wu, J.-L.; Liang, W.-B.; Zhu, R.; Xu, Z.-K.; Du, Q.; Miao, Y. Association of increased DNA methyltransferase expression with carcinogenesis and poor prognosis in pancreatic ductal adenocarcinoma. Clin. Transl. Oncol. 2012, 14, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, J.; Zhang, Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res. 2013, 23, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef]
- Wei, Y.; Xia, W.; Zhang, Z.; Liu, J.; Wang, H.; Adsay, N.V.; Albarracin, C.; Yu, D.; Abbruzzese, J.L.; Mills, G.B.; et al. Loss of Trimethylation at Lysine 27 of Histone H3 Is a Predictor of Poor Outcome in Breast, Ovarian, and Pancreatic Cancers. Mol. Carcinog. 2008, 47, 701–706. [Google Scholar] [CrossRef]
- Schneider, G.; Krämer, O.H.; Schmid, R.M.; Saur, D. Acetylation as a Transcriptional Control Mechanism—HDACs and HATs in Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Cancer 2011, 42, 85–92. [Google Scholar] [CrossRef]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2017, 66, 168–179. [Google Scholar] [CrossRef]
- Taketo, K.; Konno, M.; Asai, A.; Koseki, J.; Toratani, M.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H.; Ogawa, K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018, 52, 621–629. [Google Scholar] [CrossRef]
- He, Y.; Hu, H.; Wang, Y.; Yuan, H.; Lu, Z.; Wu, P.; Liu, D.; Tian, L.; Yin, J.; Jiang, K.; et al. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1 Methylation. Cell. Physiol. Biochem. 2018, 48, 838–846. [Google Scholar] [CrossRef]
- Martínez-Useros, J.; Georgiev-Hristov, T.; Fernandez-Aceñero, M.J.; Borrero-Palacios, A.; Indacochea, A.; Guerrero, S.; Li, W.; Cebrián, A.; Del Pulgar, T.G.; Puime-Otin, A.; et al. UNR/CDSE1 expression as prognosis biomarker in resectable pancreatic ductal adenocarcinoma patients: A proof-of-concept. PLoS ONE 2017, 12, e0182044. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Martinez-Useros, J.; Garcia-Carbonero, N.; Fernandez-Aceñero, M.J.; Ortega-Medina, L.; Garcia-Botella, S.; Perez-Aguirre, E.; Diez-Valladares, L.; Garcia-Foncillas, J. The Prognosis Value of PIWIL1 and PIWIL2 Expression in Pancreatic Cancer. J. Clin. Med. 2019, 8, 1275. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Trial | Year | Country/Region | N | Regimens | Survival Outcomes |
|---|---|---|---|---|---|
| Bakkevold [13] | 1993 | Norway | 47 | AMF (5-FU, doxorubicin, mitomycin C) (n = 23) vs. observation (n = 24) | mOS 23 mo vs. 11 mo (p = 0.02); 2-year survival 43% vs. 32%; 5-year survival 4% vs. 8% |
| Takada [14] | 2002 | Japan | 158 | MF (5-FU and mitomycin C) (n = 81) vs. observation (n = 77) | 5-year OS 17.8% vs. 26.6% in patients with curative resection (p = 0.4544); 5-year DFS 8.6% vs. 7.8% (p = 0.8372) |
| ESPAC-1 [15] | 2004 | Europe | 289 | 5-FU/folinic acid (with and without chemoradiotherapy) (n = 147) vs. no chemotherapy (observation and chemoradiotherapy) (n = 142) | mOS 20.1 mo vs. 15.5 mo (p = 0.009); 2-year estimated OS 40% vs. 30%; 5-year estimated OS 21% vs. 8%; mDFS 15.3 mo vs. 9.4 mo (p = 0.02); 1-year DFS 58% vs. 43% |
| JSAP [16] | 2006 | Japan | 89 | 5-FU/cisplatin (n = 45) vs. observation (n = 44) | mOS 12.5 mo vs. 15.8 mo; 5-year OS 26.4% vs. 14.9% |
| CONKO-001 [17,18] | 2007 and 2013 | Germany and Austria | 368 | Gemcitabine 6 cycles (n = 179) vs. observation (n = 175) | mOS 22.8 mo vs. 20.2 mo (p = 0.01); 5-year OS 20.7% vs. 10.4%; 10-year OS 12.2% vs. 7.7%; mDFS 13.4 mo vs. 6.9 mo (p < 0.001); 3-year DFS 23.5% vs. 7.5%; 5-year DFS 16.5% vs. 5.5% |
| Yoshitomi [19] | 2008 | Japan | 100 | Gemcitabine and UFT (uracil/tegafur) (n = 50) vs. gemcitabine (n = 49) | mOS 21.2 mo vs. 29.8mo (p = 0.28); 1-year OS 80.0% vs. 85.7%; 3-year OS 30.4% vs. 46.9%; mDFS 12.3 mo vs. 12.0 mo (p = 0.67); 1-year DFS 50.0% vs. 49.0%; 3-year DFS 17.7% vs. 21.6% |
| ESPAC-1 plus [20] | 2009 | Europe | 192 | 5-FU/folinic acid (n = 97) vs. observation (n = 95) | mOS 24.0 mo vs. 12.8 mo; 2-year OS 49% vs. 28%; 5-year OS 24% vs. 14% |
| ESPAC-3 v1 [20] | 2009 | Europe | 122 | 5-FU/folinic acid (n = 61) vs. observation (n = 61) | mOS 25.9 mo vs. 20.3 mo; 2-year OS 54% vs. 48%; 5-year OS 20% vs. 20% |
| JSAP-02 [21] | 2009 | Japan | 119 | Gemcitabine 3 cycles (n = 58) vs. observation (n = 60) | mOS 22.3 mo vs. 18.4 mo (p = 0.19); mDFS 11.4 mo vs. 5.0 mo (p = 0.01) |
| ESPAC-3 v2 [22] | 2010 | International | 1088 | 5-FU/folinic acid (n = 551) vs. gemcitabine (n = 537) for 6 mo | mOS 23.0 mo vs. 23.6 mo (p = 0.39); estimated 2-year OS 48.1% vs. 49.1%; mPFS 14.1 mo vs. 14.3 mo (p = 0.53); estimated 2-year PFS 30.7% vs. 29.6% |
| RTOG 97-04 [23,24] | 2011 | USA and Canada | 451 | 5-FU (n = 230) vs. gemcitabine (n = 221), both with (before and after) CRT (5-FU and 50.4 Gy) | For pancreatic head tumors, mOS 17.1 mo vs. 20.5 mo (p = 0.12); 5-year OS 18% vs. 22% |
| PACT-7 [25] | 2012 | Italy and Switzerland | 102 | Gemcitabine (n = 51) vs. PEFG (cisplatin, epirubicin, 5-FU, gemcitabine) (n = 49), both followed by chemoradiation (5-FU and 54–60 Gy) | mOS 24.8 mo vs. 28.9 mo; mDFS 11.7 mo vs. 15.2 mo; 1-year DFS 49.0% vs. 69.4% |
| Shimoda [26] | 2015 | Japan | 57 | S-1 (n = 29) vs. gemcitabine (n = 28) | mOS 21.5 mo vs. 18.0 mo (p = 0.293); 2-year OS 46% vs. 38%; mDFS 14.6 mo vs. 10.5 mo (p = 0.188); 2-year DFS 41% vs. 18% |
| JASPAC 01 [27] | 2016 | Japan | 385 | S-1 (n = 187) vs. gemcitabine (n = 190) | mOS 46.5 mo vs. 25.5 mo (p < 0.0001); 5-year OS 44.1% vs. 24.4%; mRFS 22.9 mo vs. 11.3 mo (p < 0.0001); 5-year RFS 33.3% vs. 16.8%; recurrence 66% vs. 78% |
| ESPAC-4 [28] | 2017 | Europe | 732 | Gemcitabine and capecitabine (n = 364) vs. gemcitabine (n = 366) | mOS 28.0 mo vs. 25.5 mo (p = 0.032); estimated 1-year OS 84.1% vs. 80.5%; estimated 2-year OS 53.8% vs. 52.1%; in R1 patients, mOS 23.7 mo vs. 23.0 mo; in R0 patients, mOS 39.5 mo vs. 27.9 mo (p = 0.0001) |
| CONKO-005 [29] | 2017 | Germany | 436 | Gemcitabine and erlotinib (n = 219) vs. gemcitabine (n = 217) | mOS 24.5 mo vs. 26.5 mo (p = 0.61); estimated 2-year OS 53% vs. 54%; estimated 5-year OS 23% vs. 20%; mDFS 11.4 mo vs. 11.4 mo (p = 0.26); estimated 2-year DFS 25% vs. 25%; estimated 5-year DFS 12% vs. 11% |
| PACT-15 [30] | 2018 | Italy | 93 | Gemcitabine 6 cycles (n = 26) vs. PEXG (gemcitabine, cisplatin, epirubicin, capecitabine) 6 cycles (n = 30) | mOS 20.4 mo vs. 26.4 mo; 3-year OS 35% vs. 43%; 5-year OS 13% vs. 24%; mDFS 4.7 mo vs. 12.4 mo; 1-year DFS 23% vs. 50% |
| Trial | Year | Country/Region | N | Regimens | Outcomes |
|---|---|---|---|---|---|
| Neoadjuvant chemotherapy | |||||
| Palmer [36] | 2007 | UK | 50, resectable PC | Gemcitabine (n = 24) vs. gemcitabine and cisplatin (n = 26) | Resection rate 38% vs. 70%; R0 resection 75% vs. 75%; mOS 9.9 mo vs. 15.6 mo; 1-year OS 41.7% vs. 61.5% |
| Sahora [37] | 2014 | Austria | 30, 11x borderline resectable and 19x locally advanced | Gemcitabine 4 cycles and bevacizumab 3 doses (n = 11) vs. gemcitabine 4 cycles and bevacizumab 6 doses (n = 19) | resection rate 36.4% vs. 36.8% (p = 0.97) |
| PACT-15 [30] | 2018 | Italy | 93 | PEXG (gemcitabine, cisplatin, epirubicin, capecitabine) 3 cycles before and after surgery (n = 29) vs. PEXG 6 cycles after surgery | mOS 38.2 mo vs. 26.4 mo; 3-year OS 55% vs. 43%; 5-year OS 49% vs. 24%; mDFS 16.9 mo vs. 12.4 mo; 1-year DFS 66% vs. 50% |
| Neoadjuvant CRT | |||||
| E1200 [38] | 2010 | USA | 23 | CRT (gemcitabine and 50.4Gy) (n = 10) vs. chemotherapy (gemcitabine, cisplatin, 5-FU) followed by CRT (5-FU, 50.4Gy) (n = 11), both followed by surgery and gemcitabine adjuvant chemotherapy | mOS 19.4 mo vs. 13.4 mo; 1-year acturial OS 69% vs. 61%; 2-year acturial OS 32% vs. 13%; mPFS 14.2 mo vs. not given; 1-year acturial PFS 59% vs. 15%; resectability 30% vs. 18.2% |
| Golcher [39] | 2015 | Germany and Switzerland | 73 | Upfront surgery (n = 33) vs. neoadjuvant CRT (gemcitabine, cisplatin, and 55.8 Gy) followed by surgery (n = 33), both followed by gemcitabine chemotherapy | mOS 14.4 mo vs. 17.4 mo (p = 0.96); mPFS 8.7 mo vs. 8.4 mo (p = 0.95); R0 rate 48% vs. 52% (p = 0.81); pN0 rate 30% vs. 39% (p = 0.44) |
| Casadei [40] | 2015 | Italy | 38 | Upfront surgery (n = 20) vs. neoadjuvant CRT (gemcitabine) followed by surgery (n = 18), both followed by gemcitabine chemotherapy | mOS 19.5 mo vs. 22.4 mo (p = 0.973); resectability 75% vs. 61.1% (p = 0.489); R0 rate 25.0% vs. 38.9% (p = 0.489) |
| Trial | Year | Country/Region | N | Regimens | Outcomes |
|---|---|---|---|---|---|
| Burris [47] | 1997 | USA and Canada | 126 | Gemcitabine (n = 63) vs. 5-FU (n = 63) | mOS 5.65 mo vs. 4.41 mo (p = 0.0025); 1-year OS 18% vs. 2%; mPFS 2.33 mo vs. 0.92 mo (p = 0.0002); 1-year PFS 9% vs. 5%; clinical benefit response 23.8% vs. 4.8 (p = 0.0022) |
| Huguier [48] | 2001 | France | 45 | 5-FU + leucovorin + cisplatin (n = 22) vs. best supportive care (n = 23) | mOS 8.6 mo vs. 7.0 mo |
| Ducreux [49] | 2002 | France | 207 | 5-FU + cisplatin (n = 104) vs. 5-FU (n = 103) | response 12% vs. 0% (p < 0.01); 1-year OS 17% vs. 9% (p = 0.10); 1-year PFS 10% vs. 0% (p = 0.0001) |
| Colucci [50] | 2002 | Italy | 107 | Gemcitabine + cisplatin (n = 53) vs. gemcitabine (n = 54) | mOS 30 weeks vs. 20 weeks (p = 0.43); response 26.4% vs. 9.2% (p = 0.02); mTTP (time to progression) 20 weeks vs. 8 weeks (p = 0.048) |
| Scheithauer [51] | 2003 | Austria | 83 | Gemcitabine + capecitabine (2500 mg/m2 qd 1/2 weeks) (n = 41) vs. gemcitabine (high-dose intense) (n = 42) | mOS 9.5 mo vs. 8.2 mo; 1-year OS 31.8% vs. 37.2%; mPFS 5.1 mo vs. 4.0 mo; response 17% vs. 14%; clinical benefit 48.4% vs. 33%; P values not reported |
| Tempero [52] | 2003 | USA and Netherlands | 92 | dose-intense gemcitabine (n = 49) vs. fixed dose rate gemcitabine (n = 43) | mOS 5.0 mo vs. 8.0 mo (p = 0.013); 1-year OS 9% vs. 28.8% (p = 0.014); 2-year OS 2.2% vs. 18.3% (p = 0.007); mPFS 1.9 mo vs. 3.4 mo (p = 0.68) mTTF (time to treatment failure) 1.8 mo vs. 2.1 mo (p = 0.09) |
| Ducreux [53] | 2004 | France | 63 | 5-FU + oxaliplatin (n = 31) vs. 5-FU (n = 15) vs. oxaliplatin (n = 17) | mOS 9.0 mo vs. 2.4 mo vs. 3.4 mo; mPFS 4.2 mo vs. 1.5 mo vs. 2.0 mo; response 10% vs. 0% vs. 0%; stable 48% vs. 20% vs. 12% |
| Louvet (GERCOR GISCAD) [54] | 2005 | France and Italy | 326 | Gemcitabine + oxaliplatin (n = 157) vs. gemcitabine (n = 156) | mOS 9.0 mo vs. 7.1 mo (p = 0.13); 1-year OS 34.7% vs. 27.8% (p = 0.22); mPFS 5.8 mo vs. 3.7 mo (p = 0.04); response 26.8% vs. 17.3% (p = 0.04); clinical benefit 38.2% vs. 26.9% (p = 0.03) |
| Heinemann [55] | 2006 | Germany | 195 | Gemcitabine + cisplatin (n = 98) vs. gemcitabine (n = 97) | mOS 7.5 mo vs. 6.0 mo (p = 0.15); 1-year OS 25.3% vs. 24.7% (p = 0.21); mPFS 5.3 mo vs. 3.1 mo (p = 0.053); response 10.2% vs. 8.2% ns; stable 60.2% vs. 40.2% (p < 0.001) |
| Moore (NCIC CTG PA.3) [56] | 2007 | International | 569 | Gemcitabine plus erlotinib (n = 285) vs. gemcitabine plus placebo (n = 284) | mOS 6.24 mo vs. 5.91 mo (p = 0.038); 1-year OS 23% vs. 17% (p = 0.023); mPFS 3.75 mo vs. 3.55 mo (p = 0.004); control 57.5% vs. 49.2% (p = 0.07) |
| Herrmann [57] (SAKK 44/00-CECOG/PAN.1.3.001) | 2007 | Europe | 319 | Gemcitabine + capecitabine (650 mg/m2 bid po 2/3 weeks) (n = 160) vs. gemcitabine (standard dose) (n = 159) | mOS 8.4 mo vs. 7.2 mo (p = 0.234); 1-year OS 32% vs. 30%; mPFS 4.3 mo vs. 3.9 mo (p = 0.103); response 10.0% vs. 7.8%; clinical benefit 19% vs. 20% |
| Boeck [58] | 2008 | Germany | 190 | Capecitabine plus oxaliplatin (n = 61) vs. capecitabine plus gemcitabine (n = 64) vs. gemcitabine plus oxaliplatin (n = 63) | mOS 8.1 mo vs. 9.0 mo vs. 6.9 mo (p = 0.56); 1-year OS 29% vs. 33% vs. 22%; mPFS 4.2 mo vs. 5.7 mo vs. 3.9 mo (p = 0.67); 1-year PFS 8% vs. 14% vs. 8%; response 13% vs. 25% vs. 13% (p = 0.13) |
| Cunningham [59] | 2009 | UK | 533 | Gemcitabine + capecitabine (830 mg/m2 bid po 3/4 weeks) (n = 267) vs. gemcitabine (standard dose) (n = 266) | mOS 7.1 mo vs. 6.2 mo (p = 0.08); 1-year OS 24.3% vs. 22.0%; mPFS 5.3 mo vs. 3.8 mo (p = 0.004); 1-year PFS 13.9% vs. 8.4%; response 19.1% vs. 12.4% (p = 0.03) |
| Poplin (E6201) [60] | 2009 | USA | 824 | Gemcitabine (n = 275) vs. fixed dose rate gemcitabine (n = 277) vs. gemcitabine plus oxaliplatin (n = 272) | mOS 4.9 mo vs. 6.2 mo vs. 5.7 mo; 1-year OS 16% vs. 22% vs. 21%; 2-year OS 4% vs. 6% vs. 6%; mPFS 2.6 mo vs. 3.5 mo vs. 2.7 mo |
| Kulke (CALGB 89904) [61] | 2009 | USA | 245 | Gemcitabine plus cisplatin (n = 62) vs. fixed dose rate gemcitabine (n = 58) vs. gemcitabine plus docetaxel (n = 65) vs. gemcitabine plus irinotecan (n = 60) | mOS 6.7 mo vs. 6.4 mo vs. 6.4 mo vs. 7.1 mo; mTTP 4.5 mo vs. 3.3 mo vs. 4.1 mo vs. 4.0 mo; response 13% vs. 14% vs. 12% vs. 14% |
| Colucci (GIP-1) [62] | 2010 | Italy | 400 | Gemcitabine + cisplatin (n = 201) vs. gemcitabine (n = 199) | mOS 7.2 mo vs. 8.3 mo (p = 0.38); 1-year OS 30.7% vs. 34.0%; mPFS 3.8 mo vs. 3.9 mo (p = 0.80); 1-year PFS 14.5% vs. 12.8%; response 12.9% vs. 10.1% (p = 0.37); clinical benefit 15.1% vs. 23.0% (p = 0.057) |
| Dahan (FFCD 0301) [63] | 2010 | France | 202 | 5-FU/folinic acid/cisplatin followed by gemcitabine (n = 102) vs. gemcitabine followed by 5-FU/folinic acid/cisplatin (n = 100) | mOS 6.7 mo vs. 8.03 mo (p = 0.83); mPFS 3.4 mo vs. 3.5 mo (p = 0.67); response 15% vs. 19% |
| PRODIGE 4/ ACCORD 11 [64] | 2011 | France | 342 | FOLFIRINOX (n = 171) vs. gemcitabine (n = 171) | mOS 11.1 mo vs. 6.8 mo (P <0.001); 1-year OS 48.4% vs. 20.6%; mPFS 6.4 mo vs. 3.3 mo (p < 0.001); 1-year PFS 12.1% vs. 3.5%; response 31.6% vs. 9.4% (p < 0.001); 6-month degradation QoL 31% vs. 66% (p < 0.001) |
| Ozaka (JACCRO PC-01) [65] | 2012 | Japan | 112 | Gemcitabine plus S-1 (n = 53) vs. gemcitabine (n = 59) | mOS 13.7 mo vs. 8.0 mo (p = 0.035); 1-year OS 55.9% vs. 29.0%; mPFS 6.15 mo vs. 3.78 mo (p = 0.0007); response 28.3% vs. 6.8% (p = 0.005) |
| Nakai (GEMSAP) [66] | 2012 | Japan | 106 | Gemcitabine plus S-1 (n = 53) vs. gemcitabine (n = 53) | mOS 13.5 mo vs. 8.8 mo (p = 0.104); 1-year OS 52.8% vs. 30.2% (p = 0.031); mPFS 5.4 mo vs. 3.6 mo (p = 0.036); response 18.9% vs. 9.4% (p = 0.265) |
| Chao [67] | 2013 | Taiwan | 46 | Gemcitabine + ciaplatin (n = 21) vs. gemcitabine (n = 25) | mOS 7.9 mo vs. 7.7 mo (p = 0.752); 1-year OS 9.5% vs. 12%; mTTP 3.6 mo vs. 4.6 mo (p = 0.857); partial response 4.8% vs. 8% (p = 1); clinical benefit 29% vs. 36% (p = 0.592) |
| Von Hoff and Goldstein (MPACT) [68] | 2013 and 2015 | International 11 countries | 861 | Gemcitabine + nab-paclitaxel (n = 431) vs. gemcitabine (n = 430) | mOS 8.7 mo vs. 6.6 mo (p < 0.001); 1-year OS 35% vs. 22% (p < 0.001); 2-year OS 10% vs. 5%; 3-year OS 4% vs. 0%; mPFS 5.5 mo vs. 3.7 mo (p < 0.001); 1-year PFS 16% vs. 9%; response 23% vs. 7% (p < 0.001); mTTF 5.1 mo vs. 3.6 mo (p < 0.001) |
| Ueno and Okusaka (GEST) [69,70] | 2013 and 2017 | Japan and Taiwan | 834 | Gemcitabine plus S-1 (n = 275) vs. S-1 (n = 280) vs. gemcitabine (n = 277) | mOS 9.9 mo vs. 9.7 mo vs. 8.8 mo; 1-year OS 40.7% vs. 38.7% vs. 35.4%; 2-year OS 14.5% vs. 12.7% vs. 9.2%; mPFS 5.7 mo vs. 3.8 mo vs. 4.1 mo; 1-year PFS 20.3% vs. 7.2% vs. 9.1%; response 29.3% vs. 21.0% vs. 13.3% |
| Sudo [71] | 2014 | Japan | 101 | Gemcitabine plus S-1 (n = 51) vs. gemcitabine (n = 50) | mOS 8.6 mo vs. 8.6 mo (p = 0.714); mPFS 5.3 mo vs. 3.8 mo (p = 0.039); response 21.6% vs. 6% (p = 0.048) |
| Petrioli [72] | 2015 | Italy | 67 | Gemcitabine + capecitabine + oxaliplatin (n = 34) vs. gemcitabine (n = 33) | mOS 11.9 mo vs. 7.1 mo (p < 0.001); mPFS 6.8 mo vs. 3.7 mo (p < 0.001); 4-month control 79.4% vs. 45.4% (p = 0.08) |
| Wang [73] | 2015 | Taiwan | 88 | Gemcitabine plus erlotinib (n = 44) vs. gemcitabine (n = 44) | mOS 7.2 mo vs. 4.4 mo (p < 0.001); mPFS 3.8 mo vs. 2.4 mo (p < 0.001); control 64% vs. 25% (p < 0.001) |
| Lee [74] | 2017 | Korea | 214 | Gemcitabine + capecitabine (830 mg/m2 bid po 3/4 weeks) (n = 103) vs. gemcitabine (standard dose) (n = 101) | mOS 10.3 mo vs. 7.5 mo (p = 0.06); mPFS 6.2 mo vs. 5.3 mo (p = 0.08); response 43.7% vs. 17.6% (p = 0.001) |
| Trial | Year | Country/Region | N | Regimens | Outcomes |
|---|---|---|---|---|---|
| Chauffert [77] (2000-01 FFCD/SFRO) | 2008 | France | 119 | CRT (60 Gy, 5-FU/cisplatin) plus maintenance gemcitabine chemotherapy (n = 59) vs. gemcitabine chemotherapy (n = 60) | mOS 8.6 mo vs. 13 mo (p = 0.03); 1-year OS 32% vs. 53%; 1-year PFS 14% vs. 32% |
| Loehrer [78] | 2011 | USA | 74 | CRT (50.4 Gy, gemcitabine) (n = 34) vs. gemcitabine chemotherapy (n = 37) | mOS 11.1 mo vs. 9.2 mo (p = 0.017); mPFS 6.0 mo vs. 6.7 mo; response 6% vs. 5%; stable 68% vs. 35% |
| Hammel (LAP-07) [79] | 2016 | International | 449 | gemcitabine (n = 223) vs. gemcitabine + erlotinib (n = 219) | From first randomization, mOS 13.6 mo vs. 11.9 mo (p = 0.09); mPFS 7.8 mo vs. 6.5 mo (p = 0.26) |
| 269 | chemotherapy same as previously for 2 mo (n = 136) vs. CRT (54 Gy, capecitabine) (n = 133) | From first randomization, mOS 16.5 mo vs. 15.2 mo (p = 0.83); mPFS 8.4 mo vs. 9.9 mo (p = 0.06); local progression 46% vs. 32% (p = 0.03) | |||
| Li [80] | 2003 | Taiwan | 34 | CRT (50.4~61.2 Gy, gemcitabine) (n = 18) vs. CRT (50.4~61.2 Gy, 5-FU) (n = 16), both followed by gemcitabine chemotherapy | mOS 14.5 mo vs. 6.7 mo (p = 0.027); 1-y OS 56% vs. 31%; 2-y OS 15% vs. 0%; mTTP 7.1 mo vs. 2.7 mo (p = 0.019); response 50% vs. 12.5% (p = 0.005) |
| Wilkowski [81] | 2009 | Germany | 95 | CRT (50 Gy, 5-FU) (n = 30) vs. CRT (50 Gy, gemcitabine/cisplatin) (n = 32) vs. CRT (50 Gy, gemcitabine/cisplatin) followed by gemcitabine/cisplatin chemotherapy (n = 31) | mOS 9.6 mo vs. 9.3 mo vs. 7.3 mo (p = 0.61); 9-mo OS 58% vs. 52% vs. 45%; mPFS 4.0 mo vs. 5.6 mo vs. 6.0 mo (p = 0.21); response 19% vs. 22% vs. 13% |
| Mukherjee and Hurt [82,83] | 2013 and 2017 | UK | 114 | Induction chemotherapy with gemcitabine and capecitabine for 12 weeks, if no tumor progression, then chemotherapy with gemcitabine and capecitabine for another cycle; then CRT (50.4 Gy, capecitabine) (n = 36) vs. CRT (50.4 Gy, gemcitabine) (n = 38) | mOS 17.6 mo vs. 14.6 mo (p = 0.185); 1-year OS 79.2% vs. 64.2%; mPFS 12.0 mo vs. 10.4 mo (p = 0.120); 9-mo PFS 62.9% vs. 51.4%; response (26 weeks) 23% vs. 19% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunner, M.; Wu, Z.; Krautz, C.; Pilarsky, C.; Grützmann, R.; Weber, G.F. Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions. Int. J. Mol. Sci. 2019, 20, 4543. https://doi.org/10.3390/ijms20184543
Brunner M, Wu Z, Krautz C, Pilarsky C, Grützmann R, Weber GF. Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions. International Journal of Molecular Sciences. 2019; 20(18):4543. https://doi.org/10.3390/ijms20184543
Chicago/Turabian StyleBrunner, Maximilian, Zhiyuan Wu, Christian Krautz, Christian Pilarsky, Robert Grützmann, and Georg F. Weber. 2019. "Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions" International Journal of Molecular Sciences 20, no. 18: 4543. https://doi.org/10.3390/ijms20184543
APA StyleBrunner, M., Wu, Z., Krautz, C., Pilarsky, C., Grützmann, R., & Weber, G. F. (2019). Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions. International Journal of Molecular Sciences, 20(18), 4543. https://doi.org/10.3390/ijms20184543