Molecular and Supramolecular Interactions in Systems with Nitroxide-Based Radicals

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
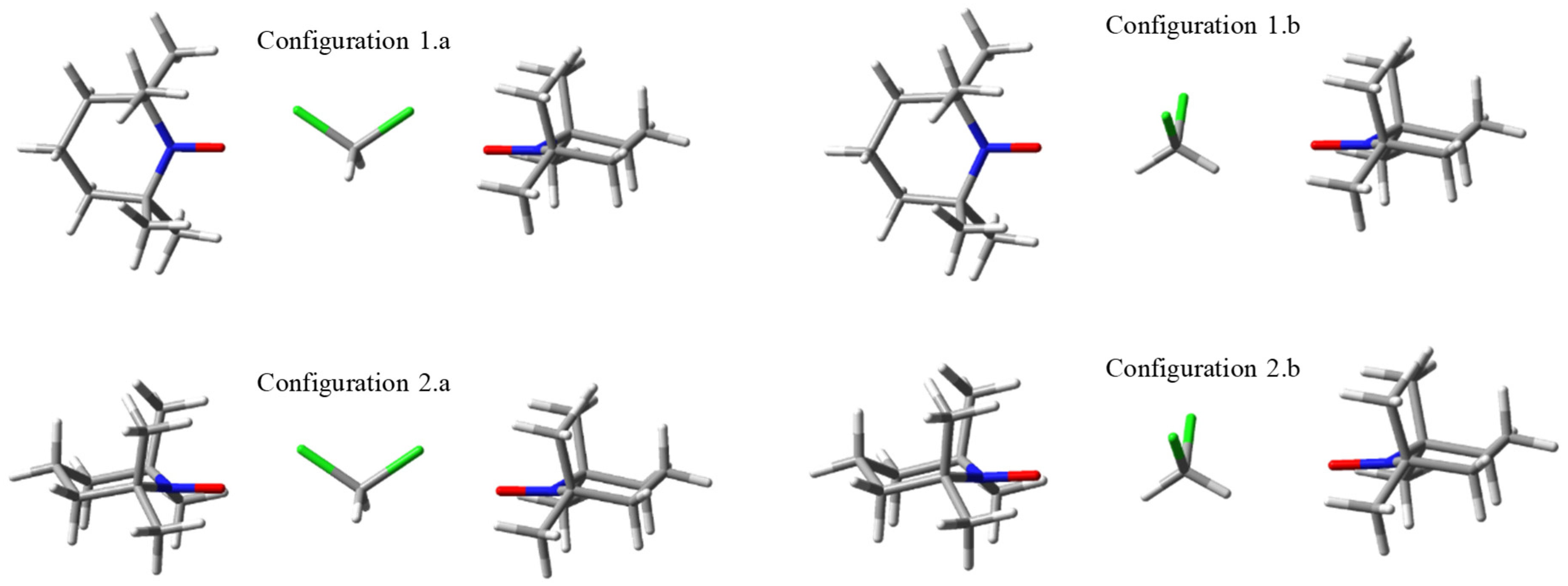
2.1. Model Geometries of TEMPO Dimeric Assemblies. Through-space vs. Solvent-mediated Exchange Coupling
2.2. Orbital-Overlap Rationalization of the Exchange Coupling in Nitroxide-Based Radical Pairs
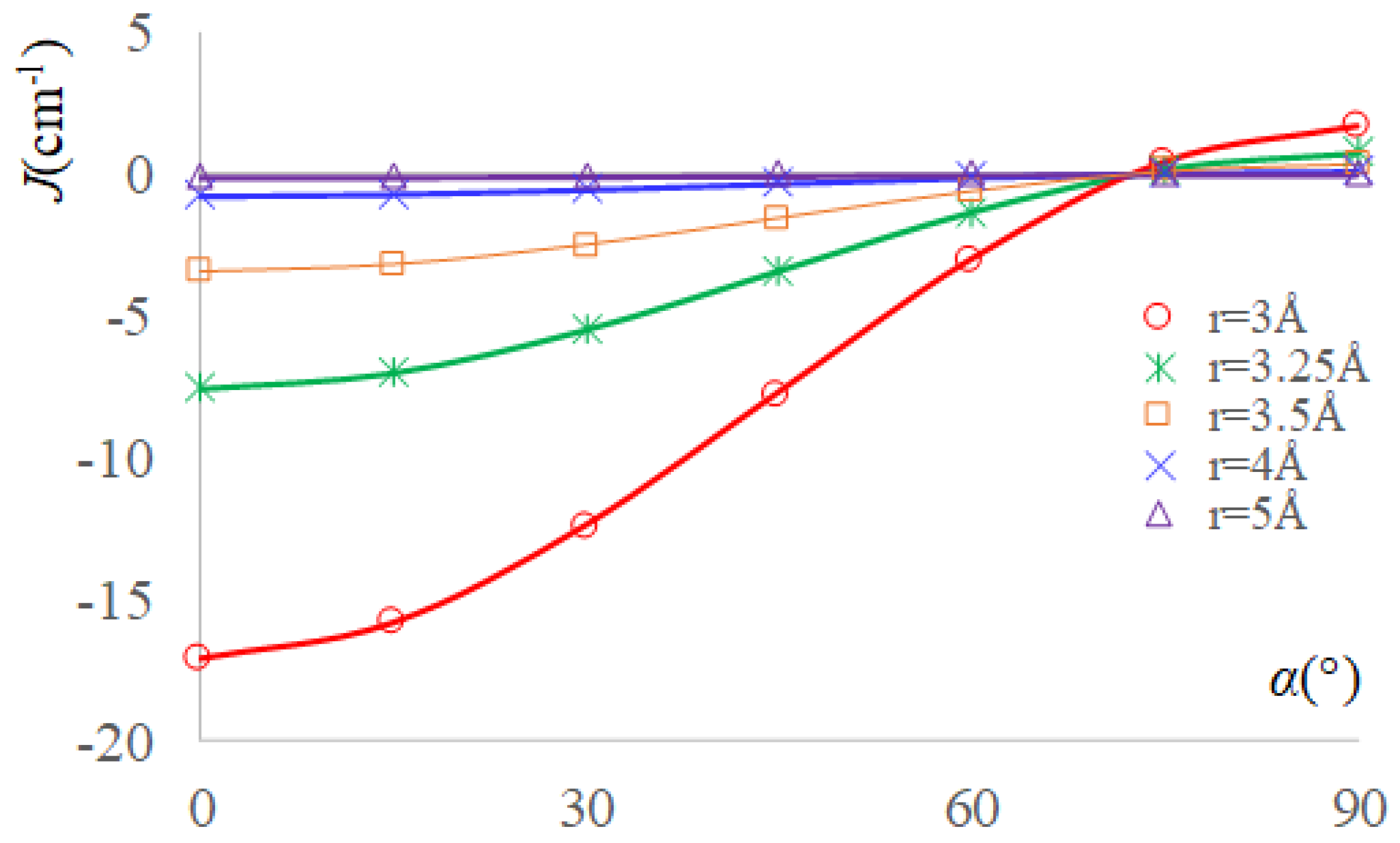
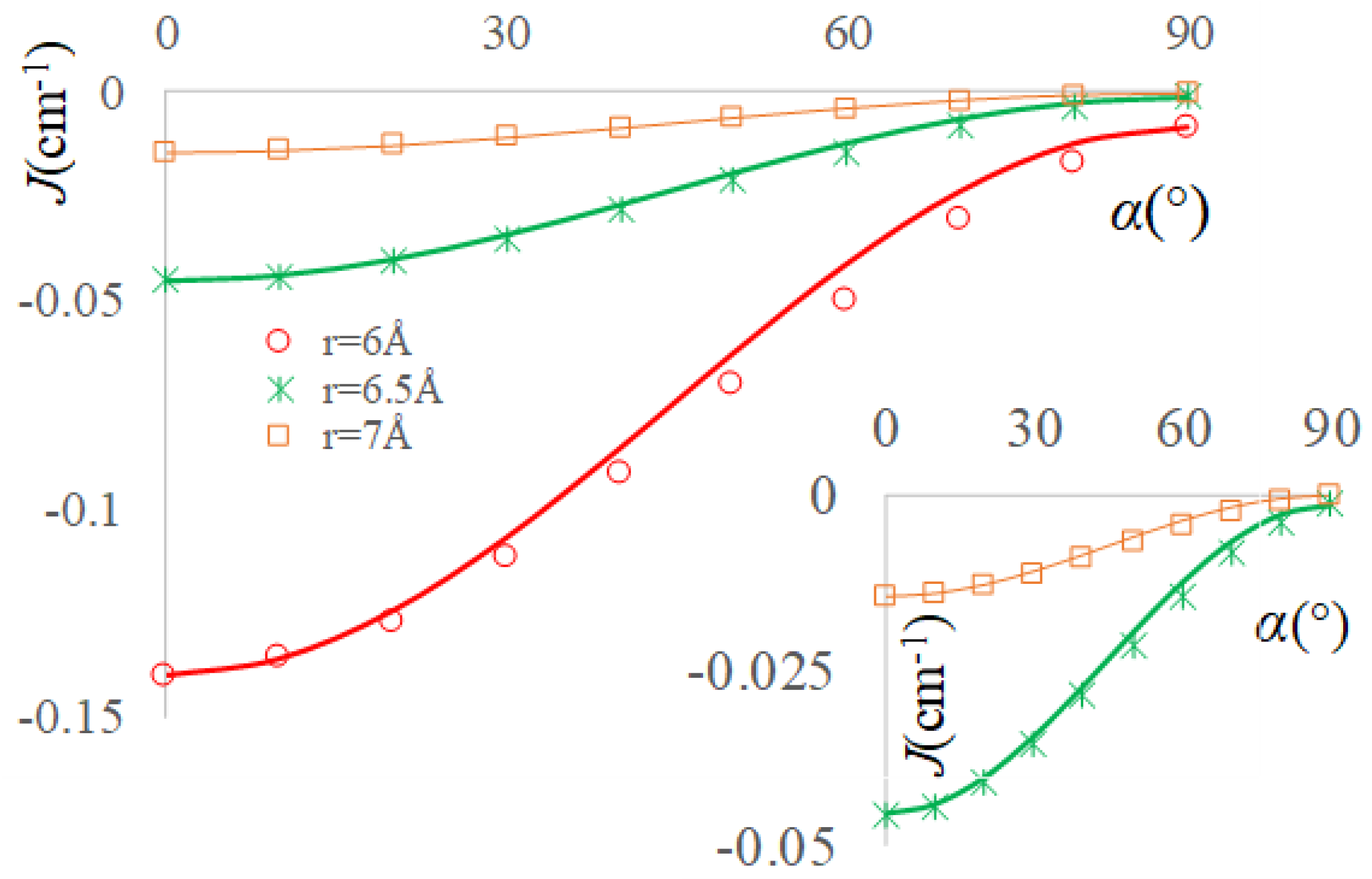
2.3. The Real Solvent Effects in Long-Range Exchange Coupling
2.4. Dipolar Coupling, Zero Field Splitting and Solvent Effects
2.5. The Basis Sets and the Hyperfine Coupling
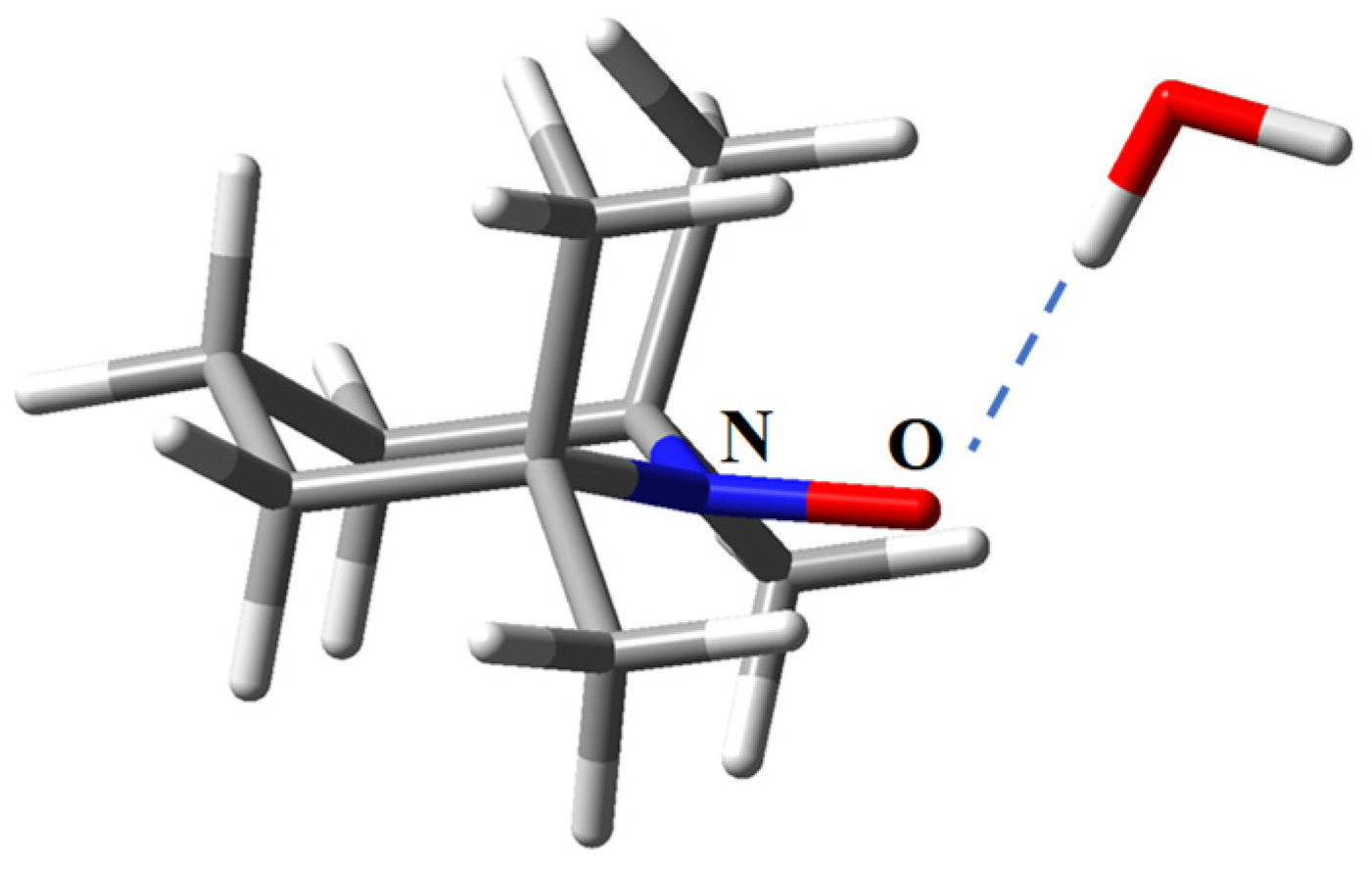
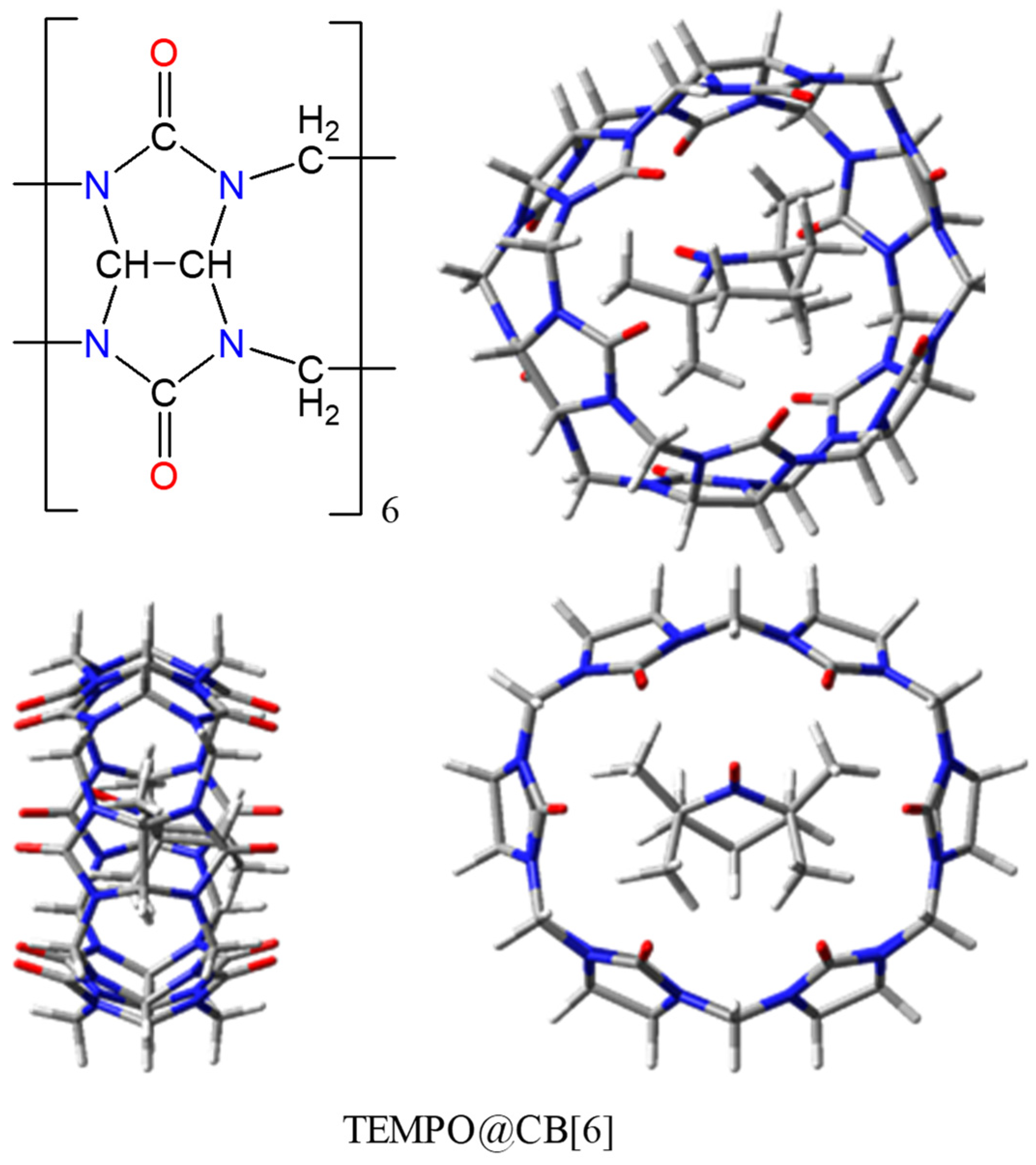
2.6. Host-guest Assemblies with TEMPO-type Radicals
3. Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ionita, G.; Caragheorgheopol, A.; Caldararu, H.; Jones, L.; Chechik, V. Inclusion complexes of cyclodextrins with nitroxide-based spin probes in aqueous solutions. Org. Biomol. Chem. 2009, 7, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Hayes Griffith, O.; Waggoner, A.S. Nitroxide free radicals: Spin labels for probing biomolecular structure. Acc. Chem. Res. 1969, 2, 17–24. [Google Scholar] [CrossRef]
- Allinger, N.L.; Burkert, U. Molecular Mechanics; American Chemical Society Publication: Washington, DC, USA, 1982. [Google Scholar]
- Mackerell, A.D. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation, 2nd ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017; p. 216. [Google Scholar]
- Benelli, C.; Gatteschi, D. Magnetism of Lanthanides in Molecular Materials with Transition-Metal Ions and Organic Radicals. Chem. Rev. 2002, 102, 2369–2388. [Google Scholar] [CrossRef] [PubMed]
- Caneschi, A.; Gatteschi, D.; Lalioti, N.; Sangregorio, C.; Sessoli, R.; Venturi, G.; Vindigni, A.; Rettori, A.; Pini, M.G.; Novak, M.A. Cobalt(II)-Nitronyl Nitroxide Chains as Molecular Magnetic Nanowires. Angew. Chem. Int. Ed. Engl. 2001, 40, 1760–1763. [Google Scholar] [CrossRef]
- Caneschi, A.; Dei, A.; Gatteschi, D.; Poussereau, S.; Sorace, L. Antiferromagnetic coupling between rare earth ions and semiquinones in a series of 1:1 complexes. Dalton Trans. 2004, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Amabilino, D.B.; Veciana, J. Magnetism, Molecules to Materials II; Miller, J.S., Drillon, M., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Volume II, pp. 1–60. [Google Scholar]
- Kollmar, C.; Kahn, O. Ferromagnetic spin alignment in molecular systems: an orbital approach. Acc. Chem. Res. 1993, 26, 259–265. [Google Scholar] [CrossRef]
- Duffy, W., Jr.; Dubach, J.F.; Pianetta, P.A.; Deck, J.F.; Strandburg, D.L.; Miedena, A.R. Antiferromagnetic Linear Chains in the Crystalline Free Radical BDPA. J. Chem. Phys. 1972, 56, 2555–2561. [Google Scholar] [CrossRef]
- Watanabe, I.; Wada, N.; Yano, H.; Okuno, T.; Awaga, K.; Ohira, S.; Nishiyama, K.; Nagamine, K. Muon-spin-relaxation study of the ground state of the two-dimensional S=1 kagoméantiferromagnet [2-(3-N-methyl-pyridium)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol-1-oxyl 3-N-oxide]BF4. Phys. Rev. B 1998, 58, 2438–2441. [Google Scholar] [CrossRef]
- Ishida, T.; Ohira, S.; Ise, T.; Nakayama, K.; Watanabe, I.; Nogami, T.; Nagamine, K. Zero-field muon spin rotation study on genuine organic ferromagnets, 4-arylmethyleneamino-2,2,6,6-tetramethylpiperidin-1-yloxyls (aryl=4-biphenylyl and phenyl). Chem. Phys. Lett. 2000, 330, 110–117. [Google Scholar] [CrossRef]
- Chiarelli, R.; Novak, M.A.; Rassat, A.; Tholence, J.L. A ferromagnetic transition at 1.48 K in an organic nitroxide. Nature 1993, 363, 147–149. [Google Scholar] [CrossRef]
- Takahashi, M.; Turek, P.; Nakazawa, Y.; Tamura, M.; Nozawa, K.; Shiomi, D.; Ishikawa, M.; Kinoshita, M. Discovery of a quasi-1D organic ferromagnet, p-NPNN. Phys. Rev. Lett. 1991, 67, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.; Ishida, T.; Nogami, T. Molecular Metamagnet [Ni(4ImNNH)2(NO3)2] (4ImNNH = 4-Imidazolyl Nitronyl Nitroxide) and the Related Compounds Showing Supramolecular H-Bonding Interactions. Inorg. Chem. 2003, 42, 7616–7625. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.G.; Lemaire, M.T.; Ohrstrom, L.; Richardson, J.F.; Thompson, L.K.; Xu, Z.Q. Strong Supramolecular-Based Magnetic Exchange in π-Stacked Radicals. Structure and Magnetism of a Hydrogen-Bonded Verdazyl Radical:Hydroquinone Molecular Solid. J. Am. Chem. Soc. 2001, 123, 7154–7159. [Google Scholar] [CrossRef] [PubMed]
- Papoutsakis, D.; Kirby, J.B.; Jackson, J.E.; Nocera, D.G. From Molecules to the Crystalline Solid: Secondary Hydrogen-Bonding Interactions of Salt Bridges and Their Role in Magnetic Exchange. Chem. Eur. J. 1999, 5, 1474–1480. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Rey, P. Structure and magnetic ordering of a ferrimagnetic helix formed by manganese(II) and a nitronyl nitroxide radical. Inorg. Chem. 1991, 30, 3936–3941. [Google Scholar] [CrossRef]
- Iwamura, H.; Inoue, K.; Koga, N.; Hayamizu, T. Magnetism: A Supramolecular Function; Kahn, O., Ed.; Kluwer: Dordrecht, The Netherlands, 1996; p. 157. [Google Scholar]
- Kahn, O. Chemistry and Physics of Supramolecular Magnetic Materials. Acc. Chem. Res. 2000, 33, 647–657. [Google Scholar] [CrossRef]
- Parmon, V.N.; Kokorin, A.I.; Jidomirov, G.M. Stable Biradicals; Nauka: Moscow, Russia, 1980. (In Russian) [Google Scholar]
- Mitchell, J.B.; Krishna, M.C.; Kuppusamy, P.; Cook, J.A.; Russo, A. Protection against Oxidative Stress by Nitroxides. Exp. Biol. Med. 2001, 226, 620–621. [Google Scholar] [CrossRef]
- Lucarini, M.; Mezzina, E. EPR investigations of organic non-covalent assemblies with spin labels and spin probes. In Electron Paramagnetic Resonance; Gilbert, B.C., Murphy, D.M., Chechik, V., Eds.; RSC Publishing: Cambridge, United Kingdom, 2011; Volume 22, p. 41. [Google Scholar]
- Junk, M.J.N.; Spiess, H.W.; Hinderberger, D. The Distribution of Fatty Acids Reveals the Functional Structure of Human Serum Albumin. Angew. Chem. Int. Ed. 2010, 49, 8755–8759. [Google Scholar] [CrossRef]
- Muravsky, V.; Gurachevskaya, T.; Berezenko, S.; Schnurr, K.; Gurachevsky, A. Fatty Acid Binding Sites of Human and Bovine Albumins: Differences Observed by Spin Probe ESR. Spectrochim. Acta A 2009, 74, 42–47. [Google Scholar] [CrossRef]
- Ionita, G.; Ariciu, A.M.; Turcu, I.M.; Chechik, V. Properties of polyethylene glycol/cyclodextrin hydrogels revealed by spin probes and spin labelling methods. Soft Matter 2014, 10, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Ionita, G.; Ariciu, A.M.; Smith, D.K.; Chechik, V. Ion exchange in alginate gels – dynamic behaviour revealed by electron paramagnetic resonance. Soft Matter 2015, 11, 8968–8974. [Google Scholar] [CrossRef] [PubMed]
- Ionita, G.; Chechik, V. Exploring polyethylene glycol/cyclodextrin hydrogels with spin probes and EPR spectroscopy. Chem. Commun. 2010, 46, 8255–8257. [Google Scholar] [CrossRef] [PubMed]
- Rogozea, A.; Matei, I.; Turcu, I.M.; Ionita, G.; Sahini, V.E.; Salifoglou, A. EPR and Circular Dichroism Solution Studies on the Interactions of Bovine Serum Albumin with Ionic Surfactants and β-Cyclodextrin. J. Phys. Chem. B 2012, 116, 14245–14253. [Google Scholar] [CrossRef] [PubMed]
- Matei, I.; Ariciu, A.M.; Neacsu, M.V.; Collauto, A.; Salifoglou, A.; Ionita, G. Cationic Spin Probe Reporting on Thermal Denaturation and Complexation–Decomplexation of BSA with SDS. Potential Applications in Protein Purification Processes. J. Phys. Chem. B 2014, 118, 11238–11252. [Google Scholar] [CrossRef]
- Constantin, M.M.; Corbu, C.G.; Tanase, C.; Codrici, E.; Mihai, S.; Popescu, I.D.; Enciu, A.-M.; Mocanu, S.; Matei, I.; Ionita, G. Spin probe method of electron paramagnetic resonance spectroscopy—A qualitative test for measuring the evolution of dry eye syndrome under treatment. Anal. Methods 2019, 11, 965–972. [Google Scholar] [CrossRef]
- Mocanu, S.; Matei, I.; Ionescu, S.; Tecuceanu, V.; Marinescu, G.; Ionita, P.; Culita, D.; Leonties, A.; Ionita, G. Complexation of β-cyclodextrin with dual molecular probes bearing fluorescent and paramagnetic moieties linked by short polyether chains. Phys. Chem. Chem. Phys. 2017, 19, 27839–27847. [Google Scholar] [CrossRef] [PubMed]
- Kahn, O. Molecular Magnetism; Wiley-VCH: Weinheim, Germany, 1993. [Google Scholar]
- Coronado, E.; Delhaès, P.; Gatteschi, D.; Miller, J.S. Molecular Magnetism: From Molecular Assemblies to the Devices; Springer: Heidelberg, Germany, 1996. [Google Scholar]
- Abe, M. Diradicals. Chem. Rev. 2013, 113, 7011–7088. [Google Scholar] [CrossRef]
- Bencini, A.; Gatteschi, D. EPR of Exchange Coupled Systems; Springer: Heidelberg, Germany, 1990; pp. 187–189. [Google Scholar]
- Pontillon, Y.; Grand, A.; Ishida, T.; Lelievre-Berna, E.; Nogami, T.; Ressouche, E.; Schweizer, J. Spin density of a ferromagnetic TEMPO derivative: Polarized neutron investigation and ab initio calculation. J. Mater. Chem. 2000, 10, 1539–1546. [Google Scholar] [CrossRef]
- Deumal, M.; Bearpark, M.J.; Robb, M.A.; Pontillon, Y.; Novoa, J.J. The Mechanism of Magnetic Interactions in the Bulk Ferromagnet para-(Methylthio)Phenyl Nitronyl Nitroxide (YUJNEW): A First Principles, Bottom-Up, Theoretical Study. Chem. Eur. J. 2004, 10, 6422–6432. [Google Scholar] [CrossRef]
- Pontillon, Y.; Caneschi, A.; Gatteschi, D.; Grand, A.; Ressouche, E.; Sessoli, R.; Schweizer, J. Experimental Spin Density in a Purely Organic Free Radical: Visualisation of the Ferromagnetic Exchange Pathway in p-(Methylthio)phenyl Nitronyl Nitroxide, Nit(SMe)Ph. Chem. Eur. J. 1999, 5, 3616–3624. [Google Scholar] [CrossRef]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; VCH: Berlin, Germany, 2001. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Houriez, C.; Ferré, N.; Siri, D.; Masella, M. Further Insights into the Environmental Effects on the Computed Hyperfine Coupling Constants of Nitroxides in Aqueous Solution. J. Phys. Chem. B 2009, 113, 15047–15056. [Google Scholar] [CrossRef] [PubMed]
- Houriez, C.; Ferré, N.; Masella, M.; Siri, D. Prediction of nitroxide hyperfine coupling constants in solution from combined nanosecond scale simulations and quantum computations. J. Chem. Phys. 2008, 128, 244504. [Google Scholar] [CrossRef] [PubMed]
- Houriez, C.; Masella, M.; Ferre, N. Structural and atoms-in-molecules analysis of hydrogen-bond network around nitroxides in liquid water. J. Chem. Phys. 2010, 133, 124508. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; di Matteo, A.; Barone, V. Effective modeling of intrinsic and environmental effects on the structure and electron paramagnetic resonance parameters of nitroxides by an integrated quantum mechanical/molecular mechanics/polarizable continuum model approach. Theor. Chem. Acc. 2000, 104, 273–279. [Google Scholar] [CrossRef]
- Rinkevicius, Z.; Frecus, B.; Murugan, N.A.; Vahtras, O.; Kongsted, J.; Ågren, H. Encapsulation Influence on EPR Parameters of Spin-Labels: 2,2,6,6-Tetramethyl-4-methoxypiperidine-1-oxyl in Cucurbit[8]uril. J. Chem. Theory Comput. 2012, 8, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Frecus, B.; Rinkevicius, Z.; Murugan, N.A.; Vahtras, O.; Kongsted, J.; Agren, H. EPR spin Hamiltonian parameters of encapsulated spin-labels: Impact of the hydrogen bonding topology. Phys. Chem. Chem. Phys. 2013, 15, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, O.; Pavone, M.; De Angelis, F.; Barone, V. Solvent Effects on the UV (n→ π*) and NMR (13C and 17O) Spectra of Acetone in Aqueous Solution. An Integrated Car-Parrinello and DFT/PCM Approach. J. Phys. Chem. B 2005, 109, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cimino, P.; Pedone, A. An integrated computational protocol for the accurate prediction of EPR and PNMR parameters of aminoxyl radicals in solution. Magn. Reson. Chem. 2010, 48, S11–S22. [Google Scholar] [CrossRef]
- Hay, P.J.; Thibeault, J.C.; Hoffmann, R. Orbital interactions in metal dimer complexes. J. Am. Chem. Soc. 1975, 97, 4884–4899. [Google Scholar] [CrossRef]
- Kahn, O.; Galy, J.; Journaux, Y.; Morgenstern-Badarau, I. Synthesis, crystal structure and molecular conformations, and magnetic properties of a copper-vanadyl (CuII-VOII) heterobinuclear complex: Interaction between orthogonal magnetic orbitals. J. Am. Chem. Soc. 1982, 104, 2165. [Google Scholar] [CrossRef]
- Willett, R.D.; Gatteschi, D.; Kahn, O. Magneto-Structural Correlations in Exchange Coupled Systems; NATO Scientific Affairs Division, Reidel Pub. Co.: Dordrecht, The Netherlands, 1985. [Google Scholar]
- Kahn, O. Magnetism of the heteropolymetallic systems. In Theoretical Approaches. Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1987; Volume 68. [Google Scholar]
- Calzado, C.J.; Cabrero, J.; Malrieu, J.P.; Caballol, R. Analysis of the magnetic coupling in binuclear complexes. I. Physics of the coupling. J. Chem. Phys. 2002, 116, 2728–2747. [Google Scholar] [CrossRef]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Malrieu, J.P.; Caballol, R.; Calzado, C.J.; de Graaf, C.; Guihery, N. Magnetic Interactions in Molecules and Highly Correlated Materials: Physical Content, Analytical Derivation, and Rigorous Extraction of Magnetic Hamiltonians. Chem. Rev. 2014, 114, 429–492. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, E.; Alemany, P.; Alvarez, S.; Cano, J. Structural Modeling and Magneto− Structural Correlations for Hydroxo-Bridged Copper (II) Binuclear Complexes. Inorg. Chem. 1997, 36, 3683–3688. [Google Scholar] [CrossRef] [PubMed]
- Cano, J.; Alemany, P.; Alvarez, S.; Verdaguer, M.; Ruiz, E. Exchange Coupling in Oxalato-Bridged Copper(II) Binuclear Compounds: A Density Functional Study. Chem. Eur. J. 1998, 4, 476–484. [Google Scholar] [CrossRef]
- Ruiz, E.; Rodriguez-Fortea, A.; Alvarez, S.; Verdaguer, M. Is It Possible to Get High TC Magnets with Prussian Blue Analogues? A Theoretical Prospect. Chem. Eur. J. 2005, 11, 2135–2144. [Google Scholar] [CrossRef]
- Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Noodleman, L.; Davidson, E.R. Ligand spin polarization and antiferromagnetic coupling in transition metal dimers. Chem. Phys. 1986, 109, 131–143. [Google Scholar] [CrossRef]
- Ruiz, E.; Cano, J.; Alvarez, S.; Alemany, P. Broken symmetry approach to calculation of exchange coupling constants for homobinuclear and heterobinuclear transition metal complexes. J. Comp. Chem. 1999, 20, 1391–1400. [Google Scholar] [CrossRef]
- Bencini, A.; Totti, F.; Daul, C.A.; Doclo, K.; Fantucci, P.; Barone, V. Density Functional Calculations of Magnetic Exchange Interactions in Polynuclear Transition Metal Complexes. Inorg. Chem. 1997, 36, 5022–5030. [Google Scholar] [CrossRef]
- Mitani, M.; Mori, H.; Takano, Y.; Yamaki, D.; Yoshioka, Y.; Yamaguchi, K. Density functional study of intramolecular ferromagnetic interaction through m-phenylene coupling unit (I): UBLYP, UB3LYP and UHF calculation. J. Chem. Phys. 2000, 113, 4035–4050. [Google Scholar] [CrossRef]
- Onishi, T.; Takano, Y.; Kitagawa, Y.; Kawakami, T.; Yoshioka, Y.; Yamaguchi, K. Theoretical study of the magnetic interaction for M–O–M type metal oxides. Comparison of broken-symmetry approaches. Polyhedron 2001, 20, 1177–1184. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pang, X.; Jin, W.J. Exploring the halogen bond specific solvent effects in halogenated solvent systems by ESR probe. New J. Chem. 2015, 39, 5477–5483. [Google Scholar] [CrossRef]
- Zhao, C.; Lu, Y.; Zhu, Z.; Liu, H. Theoretical Exploration of Halogen Bonding Interactions in the Complexes of Novel Nitroxide Radical Probes and Comparison with Hydrogen Bond. Phys. Chem. A 2018, 122, 5058–5068. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.W. Antiferromagnetism. Theory of Superexchange Interaction. Phys. Rev. 1950, 79, 350. [Google Scholar] [CrossRef]
- Cimpoesu, F.; Ferbinteanu, M. Magnetic Anisotropy in Case Studies. In Quantum Nanosystems. Structure, Properties, and Interactions; Chapter 7; Apple Academics: Ontario, ON, Canada; Florida, NJ, USA, 2014. [Google Scholar]
- Maretti, L.; Ferbinteanu, M.; Cimpoesu, F.; Islam, S.S.M.; Ohba, Y.; Kajiwara, T.; Yamashita, M.; Yamauchi, S. Spin Coupling in the supramolecular structure of a new tetra (quinolinetempo) yttrium(iii)complex. Inorg. Chem. 2007, 46, 660–669. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev.—Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Rolfe, R.E.; Sales, K.D.; Utley, J.H.P. An electron spin resonance study of conformational inversion in 4-alkyl-piperidine nitroxides. J. Chem. Soc. 1973, 2, 1171–1177. [Google Scholar] [CrossRef]
- Kumara Dhas, M.; Jawahar, A.; Benial, A.M.F. Electron spin resonance spectroscopy studies on reduction process of nitroxyl radicals used in molecular imaging. Eur. J. Biophys. 2014, 2, 1–6. [Google Scholar] [CrossRef]
- Barone, V. Recent Advances in Density Functional Methods, Part I; Chong, D.P., Ed.; World Scientific Publ. Co.: Singapore, 1996. [Google Scholar]
- Putz, M.V.; Cimpoesu, F.; Ferbinteanu, M. Structural Chemistry, Principles, Methods, and Case Studies; Springer: Cham, Switzerland, 2018; pp. 170–186. [Google Scholar]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Baseggio, O.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; et al. ADF2019, SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands.
- Holzwartha, N.A.W.; Tackettb, A.R.; Matthewsa, G.E. A Projector Augmented Wave (PAW) code for electronic structure calculations, Part I: Atompaw for generating atom-centered functions. Comput. Phys. Comm. 2001, 135, 329–347. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Leininger, T.; Stoll, H.; Werner, H.-J.; Savin, A. Combining long-range configuration interaction with short-range density functionals. Chem. Phys. Lett. 1997, 275, 151–160. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. Calculation of Small Molecular Interactions by Differences of Separate Total Energies—Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–556. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef]
- Van Leeuwen, R.; Baerends, E.J. Exchange-correlation potential with correct asymptotic behaviour. Phys. Rev. A 1994, 49, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T.; Kamiya, M.; Hirao, K. Regional self-interaction correction of density functional theory. J. Comput. Chem. 2003, 24, 1592–1598. [Google Scholar] [CrossRef]
- Kohn, W.; Meir, Y.; Makarov, D.E. van der Waals Energies in Density Functional Theory. Phys. Rev. Lett. 1998, 80, 4153–4156. [Google Scholar] [CrossRef] [Green Version]
- Mourik, T.V.; Gdanitz, R.J. A critical note on density functional theory studies on rare-gas dimers. J. Chem. Phys. 2002, 116, 9620–9623. [Google Scholar] [CrossRef]
- Andersson, Y.; Langreth, D.C.; Lundqvist, B.I. van der Waals Interactions in Density-Functional Theory. Phys. Rev. Lett. 1996, 76, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Tawada, T.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Ionita, G.; Madalan, A.M.; Ariciu, A.M.; Medvedovici, A.; Ionita, P. Synthesis of novel TEMPO stable free (poly)radical derivatives and their host–guest interaction with cucurbit[6]uril. New J. Chem. 2016, 40, 503–511. [Google Scholar] [CrossRef]
- Ionita, G.; Meltzer, V.; Pincu, E.; Chechik, V. Inclusion complexes of cyclodextrins with biradicals linked by a polyether chain—An EPR study. Org. Biomol. Chem. 2007, 5, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.M. Electronic Structure Basic Theory and Practical Methods; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Bottin, F.; Leroux, S.; Knyazev, A.; Zerah, G. Large scale ab initio calculations based on three levels of parallelization. Comput. Mat. Sci. 2008, 42, 329–336. [Google Scholar] [CrossRef]
- Gonze, X.; Amadon, B.; Anglade, P.-M.; Beuken, J.-M.; Bottin, F.; Boulanger, P.; Bruneval, F.; Caliste, D.; Caracas, R.; Cote, M.; et al. ABINIT: First-principles approach of materials and nanosystem properties. Comput. Phys. Comm. 2009, 180, 2582–2615. [Google Scholar] [CrossRef]
- Gonze, X.; Rignanese, G.-M.; Verstraete, M.; Beuken, J.-M.; Pouillon, Y.; Caracas, R.; Jollet, F.; Torrent, M.; Zerah, G.; Mikami, M.; et al. A brief introduction to the ABINIT Software Package. Z. Kristallogr. 2005, 220, 558–562. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. I. The transition state method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Von Hopffgarten, M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Baerends, E.J. Pauli Repulsion Effects in Scattering from and Catalysis by Surfaces. In Cluster Models for Surface and Bulk Phenomena; NATO ASI Series (Series B: Physics); Pacchioni, G., Bagus, P.S., Parmigiani, F., Eds.; Springer: Boston, MA, USA, 1992; Volume 283. [Google Scholar]
- Bickelhaupt, F.M.; Nibbering, N.M.; van Wezenbeek, E.M.; Baerends, E.J. The Central Bond in the Three CN Dimers NC-CN, CN-CN and CN-NC: Electron Pair Bonding and Pauli Repulsion Effects. J. Phys. Chem. 1992, 96, 4864–4873. [Google Scholar] [CrossRef]
- Jensen, J.H.; Gordon, M.S. An approximate formula for the intermolecular Pauli repulsion between closed shell molecules. Mol. Phys. 1996, 89, 1313–1325. [Google Scholar] [CrossRef]
- Rackers, J.A.; Ponder, J.W. Classical Pauli repulsion: An anisotropic, atomic multipole model. J. Chem. Phys. 2019, 150, 084104. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basis/AB | TEMPO@CB[6] | TEMPO@ β -CD | ||
|---|---|---|---|---|
| ∆E (kcal/mol) | ∆Ecorr (kcal/mol) | ∆E (kcal/mol) | ∆Ecorr (kcal/mol) | |
| 6-31G | 3.91 | 24.55 | −36.51 | −25.09 |
| 6-31+G | 6.33 | 23.61 | −34.98 | −25.75 |
| 6-31++G | 6.06 | 23.62 | −34.72 | −25.73 |
| 6-31G* | 4.15 | 24.26 | −34.75 | −23.71 |
| 6-31+G* | 13.75 | 22.76 | −29.39 | −24.52 |
| 6-31++G* | 13.65 | 22.77 | −29.45 | −24.51 |
| 6-31G** | 2.64 | 22.99 | −35.12 | −24.33 |
| 6-31+G** | 12.63 | 21.53 | −29.53 | −25.17 |
| 6-31++G** | 12.56 | 21.53 | −29.53 | −25.15 |
| 6-311G | 4.73 | 23.73 | −33.60 | −25.00 |
| 6-311+G | 4.76 | 24.34 | −34.91 | −25.24 |
| 6-311++G | 4.86 | 24.46 | −35.17 | −25.23 |
| 6-311G* | 5.58 | 22.83 | −32.30 | −24.19 |
| 6-311+G* | 13.09 | 23.19 | −30.16 | −24.54 |
| 6-311++G* | 13.21 | 23.25 | −30.29 | −24.52 |
| 6-311G** | 5.14 | 22.07 | −32.68 | −24.91 |
| 6-311+G** | 13.1 | 22.1 | −29.70 | −25.23 |
| 6-311++G** | 13.17 | 22.13 | −29.64 | −25.22 |
| ecut (a.u.) | EA (a.u.) | EB (a.u.) | EAB (a.u.) | ∆E (kcal/mol) |
|---|---|---|---|---|
| 10 | −89.752721 | −666.521144 | −756.272743 | 0.704 |
| 15 | −91.420119 | −682.677029 | −774.078638 | 11.615 |
| 20 | −91.955313 | −688.117167 | −780.046476 | 16.318 |
| 25 | −92.127097 | −689.949014 | −782.046661 | 18.480 |
| 30 | −92.182236 | −690.565848 | −782.717044 | 19.477 |
| … | … | … | … | … |
| ∞ | −92.208300 | −690.879000 | −783.055000 | 20.269 |
| ecut (a.u.) | EA (a.u.) | EB (a.u.) | EAB (a.u.) | ∆E (kcal/mol) |
|---|---|---|---|---|
| 10 | −89.808568 | −843.193164 | −933.080225 | −49.255 |
| 15 | −91.429465 | −864.977950 | −956.457872 | −31.662 |
| 20 | −92.003204 | −874.457998 | −966.508544 | −29.707 |
| 25 | −92.155612 | −877.393910 | −969.588669 | −24.565 |
| 30 | −92.187830 | −878.113239 | −970.339341 | −24.016 |
| 35 | −92.193473 | −878.238044 | −970.469690 | −23.954 |
| 40 | −92.195163 | −878.253100 | −970.486430 | −23.950 |
| … | … | … | … | … |
| ∞ | −92.213100 | −878.724000 | −970.972000 | −21.900 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buta, M.C.; Toader, A.M.; Frecus, B.; Oprea, C.I.; Cimpoesu, F.; Ionita, G. Molecular and Supramolecular Interactions in Systems with Nitroxide-Based Radicals. Int. J. Mol. Sci. 2019, 20, 4733. https://doi.org/10.3390/ijms20194733
Buta MC, Toader AM, Frecus B, Oprea CI, Cimpoesu F, Ionita G. Molecular and Supramolecular Interactions in Systems with Nitroxide-Based Radicals. International Journal of Molecular Sciences. 2019; 20(19):4733. https://doi.org/10.3390/ijms20194733
Chicago/Turabian StyleButa, Maria Cristina, Ana Maria Toader, Bogdan Frecus, Corneliu I. Oprea, Fanica Cimpoesu, and Gabriela Ionita. 2019. "Molecular and Supramolecular Interactions in Systems with Nitroxide-Based Radicals" International Journal of Molecular Sciences 20, no. 19: 4733. https://doi.org/10.3390/ijms20194733
APA StyleButa, M. C., Toader, A. M., Frecus, B., Oprea, C. I., Cimpoesu, F., & Ionita, G. (2019). Molecular and Supramolecular Interactions in Systems with Nitroxide-Based Radicals. International Journal of Molecular Sciences, 20(19), 4733. https://doi.org/10.3390/ijms20194733