Histone Deacetylation Inhibitors as Therapy Concept in Sepsis



Abstract
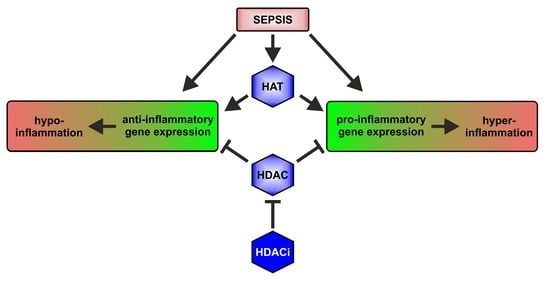
:
1. Introduction
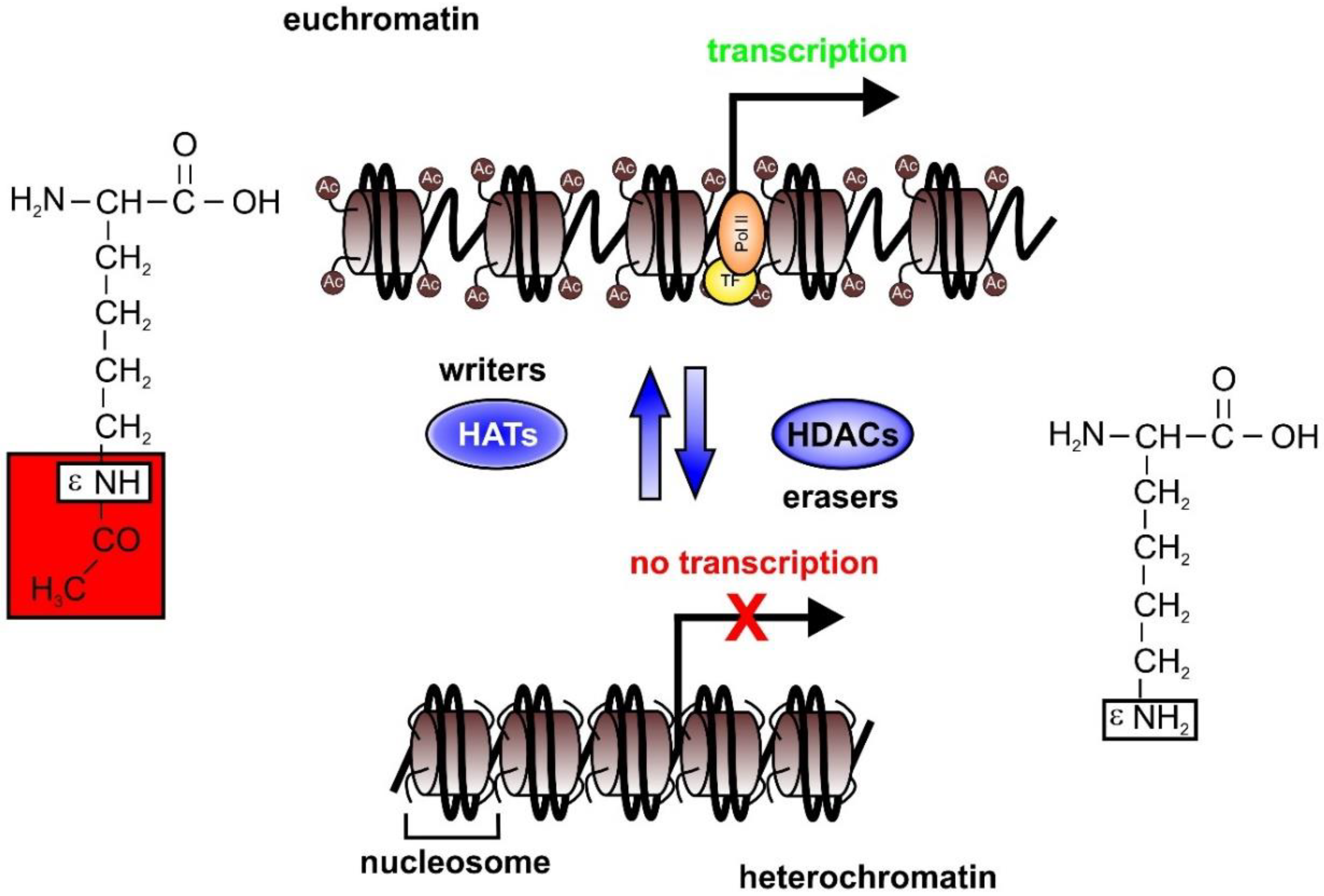
2. Epigenetics in Sepsis
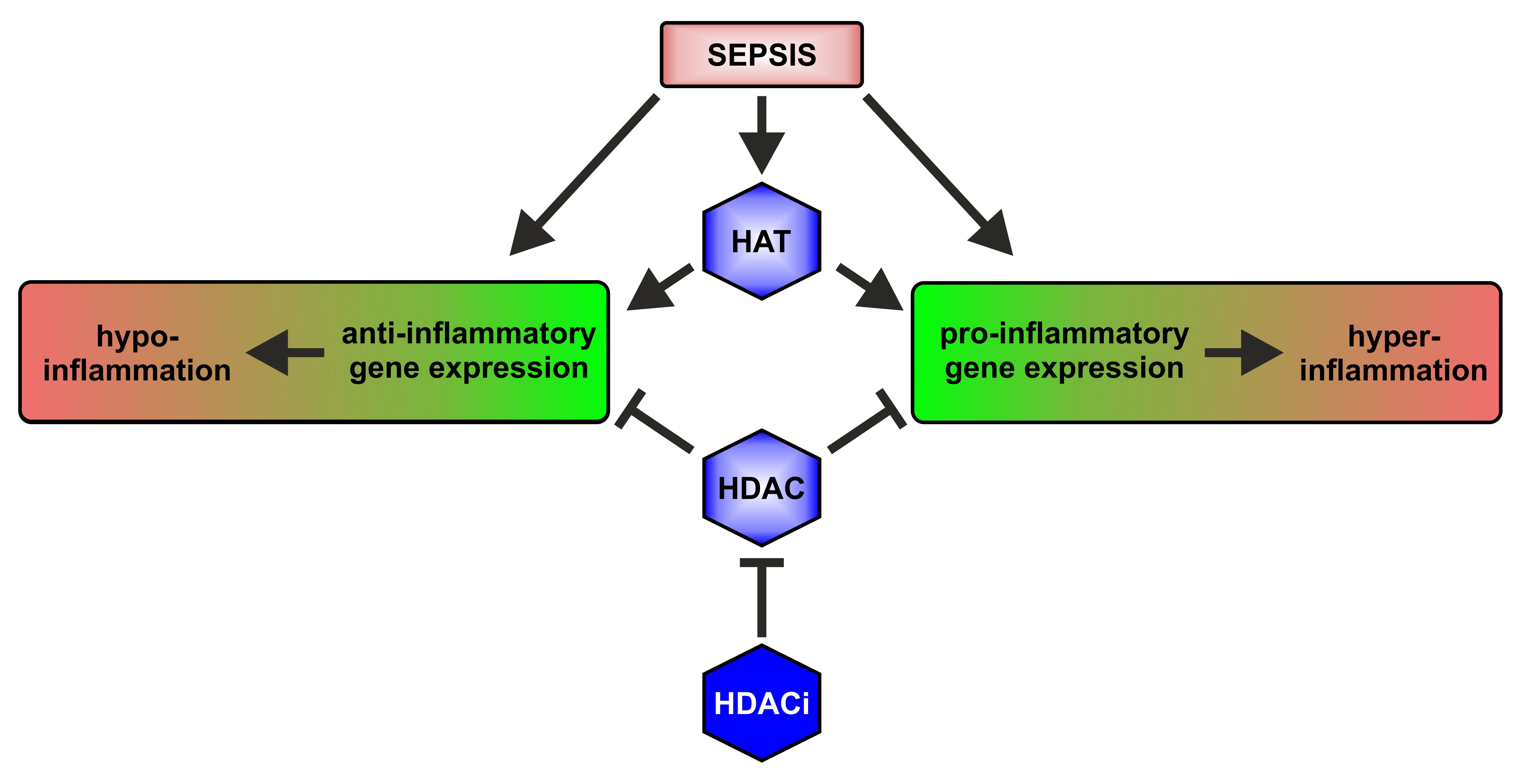
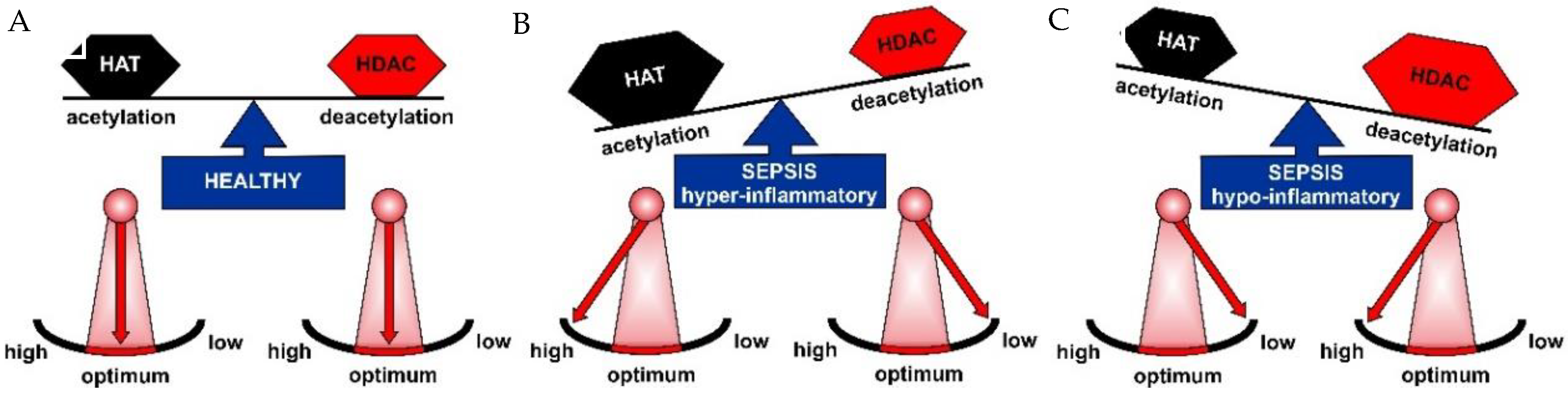
2.1. HAT and HDAC Activities in Sepsis
2.2. Polymicrobial Sepsis Mouse Models to Elucidate Epigenetic Mechanisms
2.3. Endotoxemia and LPS Treatment of Cells to Mimic Epigenetic Alterations in Sepsis
2.4. Glucocorticoids as Epigenetic Regulators in Sepsis
2.5. Role of Sirtuins in Sepsis
3. HDAC Inhibitors (HDACi) as Anti-Inflammatory Agents
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ac | acetylation |
| AKI | acute kidney injury |
| ALI | acute lung injury |
| ALT | alanine amino transferase |
| AST | aspartate amino transferase |
| BALF | bronchoalveolar lavage fluid |
| BET | bromodomain and extraterminal domain |
| BRD | bromodomain |
| CBP | CCAAT-binding protein |
| CLP | cecal ligation and puncure |
| CRP | C reactive protein |
| DC | dendritic cell |
| H | histone |
| HAT | histone acetyltransferase |
| HBO | histone acetyltransferase binding to ORC1 |
| HDAC | histone deacetylase |
| ICU | intensive care unit |
| K | lysine |
| KAT6A | lysine acetyl transferase 6A |
| LPS | lipopolysaccharide |
| MΦ | macrophage |
| MDA | malondialdehyde |
| MOF | males absent on the first |
| MPO | myeloperoxidase |
| NAD | nicotinamide adenine dinucleotide |
| SAHA | suberoylanilide hydroxamic acid |
| SAE | sepsis associated encephalopathy |
| SIRT | silent information regulator |
| TLR | toll-like receptor |
| TSA | TrichostatinA |
| TubA | Tubastatin A |
References
- Rhee, C.; Dantes, R.; Epstein, L.; Murphy, D.J.; Seymour, C.W.; Iwashyna, T.J.; Kadri, S.S.; Angus, D.C.; Danner, R.L.; Fiore, A.E.; et al. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. JAMA 2017, 318, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar-Hari, M.; Phillips, G.S.; Levy, M.L.; Seymour, C.W.; Liu, V.X.; Deutschman, C.S.; Angus, D.C.; Rubenfeld, G.D.; Singer, M. Developing a New Definition and Assessing New Clinical Criteria for Septic Shock: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Cavaillon, J.-M.; Adib-Conquy, M.; Fitting, C.; Adrie, C.; Payen, D. Cytokine cascade in sepsis. Scand. J. Infect. Dis. 2003, 35, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Bosmann, M.; Ward, P.A. The inflammatory response in sepsis. Trends Immunol. 2013, 34, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D.; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Coopersmith, C.M.; McDunn, J.E.; Ferguson, T.A. The sepsis seesaw: Tilting toward immunosuppression. Nat. Med. 2009, 15, 496–497. [Google Scholar] [CrossRef]
- Oberholzer, C.; Oberholzer, A.; Clare-Salzler, M.; Moldawer, L.L. Apoptosis in sepsis: A new target for therapeutic exploration. FASEB J. 2001, 15, 879–892. [Google Scholar] [CrossRef]
- Sun, A.; Netzer, G.; Small, D.S.; Hanish, A.; Fuchs, B.D.; Gaieski, D.F.; Mikkelsen, M.E. Association Between Index Hospitalization and Hospital Readmission in Sepsis Survivors. Crit. Care Med. 2016, 44, 478–487. [Google Scholar] [CrossRef]
- Shankar-Hari, M.; Rubenfeld, G.D. Understanding Long-Term Outcomes Following Sepsis: Implications and Challenges. Curr. Infect. Dis. Rep. 2016, 18, 37. [Google Scholar] [CrossRef] [PubMed]
- Pueschel, R.; Coraggio, F.; Meister, P. From single genes to entire genomes: The search for a function of nuclear organization. Development 2016, 143, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef]
- Voss, A.K.; Thomas, T. Histone Lysine and Genomic Targets of Histone Acetyltransferases in Mammals. BioEssays 2018, 40, e1800078. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.M.; Yao, Y.L.; Sun, J.M.; Davie, J.R.; Seto, E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997, 272, 28001–28007. [Google Scholar] [CrossRef]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef]
- Zhou, X.; Richon, V.M.; Rifkind, R.A.; Marks, P.A. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc. Natl. Acad. Sci. USA 2000, 97, 1056–1061. [Google Scholar] [CrossRef] [Green Version]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, H.Y.; Downes, M.; Ordentlich, P.; Evans, R.M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000, 14, 55–66. [Google Scholar]
- Kao, H.-Y.; Lee, C.-H.; Komarov, A.; Han, C.C.; Evans, R.M. Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J. Biol. Chem. 2002, 277, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Ptashne, M. Epigenetics: Core misconcept. Proc. Natl. Acad. Sci. USA 2013, 110, 7101–7103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddihough, G.; Zahn, L.M. Epigenetics. What is epigenetics? Introduction. Science 2010, 330, 611. [Google Scholar] [CrossRef]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Benton, C.B.; Fiskus, W.; Bhalla, K.N. Targeting Histone Acetylation: Readers and Writers in Leukemia and Cancer. Cancer J. 2017, 23, 286–291. [Google Scholar] [CrossRef]
- Cazalis, M.-A.; Lepape, A.; Venet, F.; Frager, F.; Mougin, B.; Vallin, H.; Paye, M.; Pachot, A.; Monneret, G. Early and dynamic changes in gene expression in septic shock patients: A genome-wide approach. Intensive Care Med. Exp. 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.E.; Burnham, K.L.; Radhakrishnan, J.; Humburg, P.; Hutton, P.; Mills, T.C.; Rautanen, A.; Gordon, A.C.; Garrard, C.; Hill, A.V.S.; et al. Genomic landscape of the individual host response and outcomes in sepsis: A prospective cohort study. Lancet Respir. Med. 2016, 4, 259–271. [Google Scholar] [CrossRef]
- Schaack, D.; Siegler, B.H.; Tamulyte, S.; Weigand, M.A.; Uhle, F. The immunosuppressive face of sepsis early on intensive care unit-A large-scale microarray meta-analysis. PLoS ONE 2018, 13, e0198555. [Google Scholar] [CrossRef] [PubMed]
- Warford, J.; Lamport, A.-C.; Kennedy, B.; Easton, A.S. Human Brain Chemokine and Cytokine Expression in Sepsis: A Report of Three Cases. Can. J. Neurol. Sci. 2017, 44, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Alamdari, N.; Smith, I.J.; Aversa, Z.; Hasselgren, P.-O. Sepsis and glucocorticoids upregulate p300 and downregulate HDAC6 expression and activity in skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R509–R520. [Google Scholar] [CrossRef] [Green Version]
- Ciarlo, E.; Heinonen, T.; Herderschee, J.; Fenwick, C.; Mombelli, M.; Le Roy, D.; Roger, T. Impact of the microbial derived short chain fatty acid propionate on host susceptibility to bacterial and fungal infections in vivo. Sci. Rep. 2016, 6, 37944. [Google Scholar] [CrossRef] [Green Version]
- Eskandarian, H.A.; Impens, F.; Nahori, M.-A.; Soubigou, G.; Coppée, J.-Y.; Cossart, P.; Hamon, M.A. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 2013, 341, 1238858. [Google Scholar] [CrossRef]
- Carson, W.F.; Cavassani, K.A.; Dou, Y.; Kunkel, S.L. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics 2011, 6, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Hassan, F.I.; Didari, T.; Khan, F.; Mojtahedzadeh, M.; Abdollahi, M. The Role of Epigenetic Alterations Involved in Sepsis: An Overview. Curr. Pharm. Des. 2018, 24, 2862–2869. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, Z.; Liu, B.; Zhao, T.; Li, Y.; Alam, H.B. Selective histone deacetylase 6 inhibition prolongs survival in a lethal two-hit model. J. Surg. Res. 2015, 197, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, T.; Liu, B.; Halaweish, I.; Mazitschek, R.; Duan, X.; Alam, H.B. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J. Trauma Acute Care Surg. 2015, 78, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Li, Y.; Bronson, R.T.; Liu, B.; Velmahos, G.C.; Alam, H.B. Selective histone deacetylase-6 inhibition attenuates stress responses and prevents immune organ atrophy in a lethal septic model. Surgery 2014, 156, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Lienlaf, M.; Perez-Villarroel, P.; Wang, H.-W.; Lee, C.; Woan, K.; Woods, D.; Knox, T.; Bergman, J.; Pinilla-Ibarz, J.; et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol. Immunol. 2014, 60, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Villagra, A.; Cheng, F.; Wang, H.-W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, F.; Woan, K.; Sahakian, E.; Merino, O.; Rock-Klotz, J.; Vicente-Suarez, I.; Pinilla-Ibarz, J.; Wright, K.L.; Seto, E.; et al. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J. Immunol. 2011, 186, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Jin, M. Potential Immunotherapeutics for Immunosuppression in Sepsis. Biomol. Ther. 2017, 25, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattahi, F.; Ward, P.A. Understanding Immunosuppression after Sepsis. Immunity 2017, 47, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, M.; Sudhoff, L.; Crucian, B.; Pagel, J.-I.; Sams, C.; Strewe, C.; Guo, A.; Schelling, G.; Briegel, J.; Kaufmann, I.; et al. Early immune anergy towards recall antigens and mitogens in patients at onset of septic shock. Sci. Rep. 2018, 8, 1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A.; Fossati, G.; Mascagni, P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol. Med. 2011, 17, 333–352. [Google Scholar] [CrossRef]
- Chua, M.J.; Arnold, M.S.J.; Xu, W.; Lancelot, J.; Lamotte, S.; Späth, G.F.; Prina, E.; Pierce, R.J.; Fairlie, D.P.; Skinner-Adams, T.S.; et al. Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 42–50. [Google Scholar] [CrossRef]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [PubMed]
- Clutton, G.T.; Jones, R.B. Diverse Impacts of HIV Latency-Reversing Agents on CD8+ T-Cell Function: Implications for HIV Cure. Front. Immunol. 2018, 9, 1452. [Google Scholar] [CrossRef]
- Cole, J.; Morris, P.; Dickman, M.J.; Dockrell, D.H. The therapeutic potential of epigenetic manipulation during infectious diseases. Pharmacol. Ther. 2016, 167, 85–99. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.J.; Rosengart, M.R. Bench-to-Bedside: A Translational Perspective on Murine Models of Sepsis. Surg. Infect. 2018, 19, 137–141. [Google Scholar] [CrossRef] [PubMed]
- van Lier, D.; Geven, C.; Leijte, G.P.; Pickkers, P. Experimental human endotoxemia as a model of systemic inflammation. Biochimie 2018. [Google Scholar] [CrossRef] [PubMed]
- Aung, H.T.; Schroder, K.; Himes, S.R.; Brion, K.; van Zuylen, W.; Trieu, A.; Suzuki, H.; Hayashizaki, Y.; Hume, D.A.; Sweet, M.J.; et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006, 20, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, A.; Hu, J.; Kang, J. Histone deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. Immunol. Cell Biol. 2018. [Google Scholar] [CrossRef]
- Somanath, P.; Herndon Klein, R.; Knoepfler, P.S. CRISPR-mediated HDAC2 disruption identifies two distinct classes of target genes in human cells. PLoS ONE 2017, 12, e0185627. [Google Scholar] [CrossRef]
- Wu, J.; Dong, L.; Zhang, M.; Jia, M.; Zhang, G.; Qiu, L.; Ji, M.; Yang, J. Class I histone deacetylase inhibitor valproic acid reverses cognitive deficits in a mouse model of septic encephalopathy. Neurochem. Res. 2013, 38, 2440–2449. [Google Scholar] [CrossRef]
- Pena, O.M.; Hancock, D.G.; Lyle, N.H.; Linder, A.; Russell, J.A.; Xia, J.; Fjell, C.D.; Boyd, J.H.; Hancock, R.E.W. An Endotoxin Tolerance Signature Predicts Sepsis and Organ Dysfunction at Initial Clinical Presentation. EBioMedicine 2014, 1, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Seeley, J.J.; Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2017, 101, 107–119. [Google Scholar] [CrossRef]
- Arts, R.J.W.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.-C.; Wang, S.-Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Hohenhaus, D.M.; Kelly, G.M.; Kamal, N.A.; Gupta, P.; Labzin, L.I.; Schroder, K.; Garceau, V.; Barbero, S.; Iyer, A.; et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J. Biol. Chem. 2013, 288, 25362–25374. [Google Scholar] [CrossRef]
- Zhu, H.; Shan, L.; Schiller, P.W.; Mai, A.; Peng, T. Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J. Biol. Chem. 2010, 285, 9429–9436. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wei, W.; Menconi, M.; Hasselgren, P.-O. Dexamethasone-induced protein degradation in cultured myotubes is p300/HAT dependent. Am. J. Physiol. Regul. Integr. Comp. J. Physiol. 2007, 292, R337–R344. [Google Scholar] [CrossRef]
- Walko, T.D.; Di Caro, V.; Piganelli, J.; Billiar, T.R.; Clark, R.S.B.; Aneja, R.K. Poly(ADP-ribose) polymerase 1-sirtuin 1 functional interplay regulates LPS-mediated high mobility group box 1 secretion. Mol. Med. 2015, 20, 612–624. [Google Scholar]
- Zhao, T.; Li, Y.; Liu, B.; Bronson, R.T.; Halaweish, I.; Alam, H.B. Histone deacetylase III as a potential therapeutic target for the treatment of lethal sepsis. J. Trauma Acute Care Surg. 2014, 77, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.N.; Alexander-Miller, M.; Yoza, B.K.; Vachharajani, V.; McCall, C.E. Sirtuin1 Targeting Reverses Innate and Adaptive Immune Tolerance in Septic Mice. J. Immunol. Res. 2018, 2018, 2402593. [Google Scholar] [CrossRef]
- Jia, Y.; Li, Z.; Cai, W.; Xiao, D.; Han, S.; Han, F.; Bai, X.; Wang, K.; Liu, Y.; Li, X.; et al. SIRT1 regulates inflammation response of macrophages in sepsis mediated by long noncoding RNA. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Ciarlo, E.; Heinonen, T.; Théroude, C.; Herderschee, J.; Mombelli, M.; Lugrin, J.; Pfefferlé, M.; Tyrrell, B.; Lensch, S.; Acha-Orbea, H.; et al. Sirtuin 2 Deficiency Increases Bacterial Phagocytosis by Macrophages and Protects from Chronic Staphylococcal Infection. Front. Immunol. 2017, 8, 1037. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K.-S. SIRT1/3 Activation by Resveratrol Attenuates Acute Kidney Injury in a Septic Rat Model. Oxid. Med. Cell Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, B.; Fukudome, E.Y.; Kochanek, A.R.; Finkelstein, R.A.; Chong, W.; Jin, G.; Lu, J.; deMoya, M.A.; Velmahos, G.C.; et al. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery 2010, 148, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Li, Y.; Liu, B.; Liu, Z.; Chong, W.; Duan, X.; Deperalta, D.K.; Velmahos, G.C.; Alam, H.B. Novel pharmacologic treatment attenuates septic shock and improves long-term survival. Surgery 2013, 154, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Lian, Y.; Xie, K.; Cai, S.; Wen, P. Epigenetic modulation of neuronal apoptosis and cognitive functions in sepsis-associated encephalopathy. Neurol. Sci. 2014, 35, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Zhou, Z.; Rajasingh, S.; Panda, A.; Sampath, V.; Rajasingh, J. DNMT and HDAC inhibitors together abrogate endotoxemia mediated macrophage death by STAT3-JMJD3 signaling. Int. J. Biochem. Cell Biol. 2018, 102, 117–127. [Google Scholar] [CrossRef]
- Kim, S.-J.; Baek, K.S.; Park, H.-J.; Jung, Y.H.; Lee, S.-M. Compound 9a, a novel synthetic histone deacetylase inhibitor, protects against septic injury in mice by suppressing MAPK signalling. Br. J. Pharmacol. 2016, 173, 1045–1057. [Google Scholar] [CrossRef]
- Hsing, C.-H.; Lin, C.-F.; So, E.; Sun, D.-P.; Chen, T.-C.; Li, C.-F.; Yeh, C.-H. α2-Adrenoceptor agonist dexmedetomidine protects septic acute kidney injury through increasing BMP-7 and inhibiting HDAC2 and HDAC5. Am. J. Physiol. Ren. Physiol. 2012, 303, F1443–F1453. [Google Scholar] [CrossRef]
- Zhang, L.; Jin, S.; Wang, C.; Jiang, R.; Wan, J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J. Surg. 2010, 34, 1676–1683. [Google Scholar] [CrossRef]
- Zhang, L.; Wan, J.; Jiang, R.; Wang, W.; Deng, H.; Shen, Y.; Zheng, W.; Wang, Y. Protective effects of trichostatin A on liver injury in septic mice. Hepatol. Res. 2009, 39, 931–938. [Google Scholar] [CrossRef]
- Rios, E.C.S.; de Lima, T.M.; Moretti, A.I.S.; Soriano, F.G. The role of nitric oxide in the epigenetic regulation of THP-1 induced by lipopolysaccharide. Life Sci. 2016, 147, 110–116. [Google Scholar] [CrossRef]
- Ji, M.-H.; Li, G.-M.; Jia, M.; Zhu, S.-H.; Gao, D.-P.; Fan, Y.-X.; Wu, J.; Yang, J.-J. Valproic acid attenuates lipopolysaccharide-induced acute lung injury in mice. Inflammation 2013, 36, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Steckert, A.V.; Comim, C.M.; Igna, D.M.D.; Dominguini, D.; Mendonça, B.P.; Ornell, F.; Colpo, G.D.; Gubert, C.; Kapczinski, F.; Barichello, T.; et al. Effects of sodium butyrate on aversive memory in rats submitted to sepsis. Neurosci. Lett. 2015, 595, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Georgoff, P.E.; Nikolian, V.C.; Bonham, T.; Pai, M.P.; Tafatia, C.; Halaweish, I.; To, K.; Watcharotone, K.; Parameswaran, A.; Luo, R.; et al. Safety and Tolerability of Intravenous Valproic Acid in Healthy Subjects: A Phase I Dose-Escalation Trial. Clin. Pharmacokinet. 2018, 57, 209–219. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Verner, E.; Buggy, J.J. Isoform-specific histone deacetylase inhibitors: The next step? Cancer Lett. 2009, 280, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Zhao, T.; Pan, B.; Dennahy, I.S.; Duan, X.; Williams, A.M.; Liu, B.; Lin, N.; Bhatti, U.F.; Chen, E.; et al. Protective Effect of Tubastatin A in CLP-Induced Lethal Sepsis. Inflammation 2018, 41, 2101–2109. [Google Scholar] [CrossRef]
- Zhao, T.; Li, Y.; Liu, B.; Pan, B.; Cheng, X.; Georgoff, P.; Alam, H.B. Inhibition of histone deacetylase 6 restores innate immune cells in the bone marrow in a lethal septic model. J. Trauma Acute Care Surg. 2016, 80, 34–40. [Google Scholar] [CrossRef]
- Zhao, T.; Li, Y.; Liu, B.; Halaweish, I.; Mazitschek, R.; Alam, H.B. Selective inhibition of histone deacetylase 6 alters the composition of circulating blood cells in a lethal septic model. J. Surg. Res. 2014, 190, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Tian, R.; Yang, Y.; Jiang, R.; Dai, J.; Tang, L.; Zhang, L. The SIRT1 inhibitor EX-527 suppresses mTOR activation and alleviates acute lung injury in mice with endotoxiemia. Innate Immun. 2017, 23, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Alam, H.B.; Liu, B.; Bronson, R.T.; Nikolian, V.C.; Wu, E.; Chong, W.; Li, Y. Selective Inhibition of SIRT2 Improves Outcomes in a Lethal Septic Model. Curr. Mol. Med. 2015, 15, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, J.; Samanta, S.; Rajasingh, S.; Barani, B.; Xuan, Y.-T.; Dawn, B.; Rajasingh, J. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. J. Cell Sci. 2015, 128, 3094–3105. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, J.; Malik, A.B.; Elias, H.K.; Rajasingh, S.; Simpson, A.D.; Sundivakkam, P.K.; Vogel, S.M.; Xuan, Y.-T.; Dawn, B.; Rajasingh, J. Combinatorial therapy with acetylation and methylation modifiers attenuates lung vascular hyperpermeability in endotoxemia-induced mouse inflammatory lung injury. Am. J. Pathol. 2014, 184, 2237–2249. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Feng, D.; Fang, B.; Mullican, S.E.; You, S.-H.; Lim, H.-W.; Everett, L.J.; Nabel, C.S.; Li, Y.; Selvakumaran, V.; et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell 2013, 52, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, W.; Jiao, F.; Li, X.; Zhang, H.; Wang, L.; Gong, Z. The Nephroprotective Effect of MS-275 on Lipopolysaccharide (LPS)-Induced Acute Kidney Injury by Inhibiting Reactive Oxygen Species (ROS)-Oxidative Stress and Endoplasmic Reticulum Stress. Med. Sci. Monit. 2018, 24, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Park, S.-K.; Kim, H.M.; Kang, J.S.; Yoon, Y.D.; Han, S.B.; Han, J.W.; Yang, J.S.; Han, G. Histone deacetylase inhibitor KBH-A42 inhibits cytokine production in RAW 264.7 macrophage cells and in vivo endotoxemia model. Exp. Mol. Med. 2008, 40, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.E.; Lichtenstein, R.; Lue, J.; Sawas, A.; Deng, C.; Lichtenstein, E.; Khan, K.; Atkins, L.; Rada, A.; Kim, H.A.; et al. A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 2018, 131, 397–407. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Superfamily | Family | Class | Subclass | Protein | Ref. | |
|---|---|---|---|---|---|---|
| CLASSICAL | Arginase/ deacetylase superfamily | Histone deacetylases | Class I | HDAC1, 2, 3, 8 | [16,17,18,19] | |
| Class II | a | HDAC4, 5, 7, 9 | [20,21,22] | |||
| b | HDAC6, 10 | [21,23] | ||||
| Class IV | HDAC11 | [24] | ||||
| Deoxyhypusine synthase like NAD+-binding domain superfamily | Sir2 regulators | Class III | I II III IV | SIRT1, 2, 3 SIRT4 SIRT5 SIRT6, 7 | [25] [25] [25] [26] |
| HDACi | Inhibition | Model | HDACi Effect | Ref. | |
|---|---|---|---|---|---|
| Hydroxamic acids | SAHA (Vorinostat) | pan | CLP | survival↑ | [74] |
| LPS-endotoxemia | TNF-α↓, IL-6↓ | [75] | |||
| Long-term survival following CLP | Long-term survival↑ | [75] | |||
| CLP/SAE | neuronal apoptosis↓; locomotive activity↑, H3Ac, H4Ac; nuclear HDAC4↑; Bax↓, Bcl-XL↑ | [76] | |||
| TSA | pan | LPS endotoxemia | ALI↓, apoptosis↓, inflammation↓ | [77] | |
| LPS/BMDM | DNA fragmentation↓, expression of apoptotic/pyroptotic genes↓ | [77] | |||
| CLP | survival↑, ALI↓, TNF-α↓, IL-6↓, TLR2↓, TLR4↓, MyD88↓, nuclear NF-κB↓, I-κBα↓ | [78] | |||
| Tolerance (LPS/THP-1 cells) | IL-6↑, IL-10↓ | [82] | |||
| LPS-induced ALI | IL-1β↓, TNF-α↓, lung MPO↓, PMN cells in BALF↓ | [92] | |||
| LPS-induced ALI | inflammation↓, ALI↓, survival↑ | [93,94] | |||
| CLP/SAE | neuronal apoptosis↓, locomotive activity↑, H3Ac↑, H4Ac↑, nuclear HDAC4↑; Bax↓, Bcl-XL↑ | [76] | |||
| CLP | plasma urea↓, creatinine↓, CRP↓, tubular damage↓, TNF-α↓, MCP-1↓, BMP-7↑, HDAC2/5↓, H3Ac↑ | [79] | |||
| CLP | ALI↓, neutrophil infiltra-tion↓, ICAM-1↓, E-selec-tin↓, IL-6 ↓, survival↑ | [80] | |||
| CLP | ALT/AST↓, MDA↓, MPO↓, ICAM-1↓, IL-6↓, IL-10↓ | [81] | |||
| LPS/BMDM | Cox-2↑, Cxcl2↑, Ifit2↑, Ccl2↓, Ccl7↓, Edn1↓ | [57] | |||
| Benzamides | TubA | HDAC6 | CLP | circulating monocytes↑, lymphocytes↑, granulo-cytes↓ | [89] |
| “two-hit” model | survival ↑, MPO↓, TNF-α ↓, IL-6 ↓ | [40] | |||
| CLP | survival↑, ALI↓ MPO↓, TNF-α↓, IL-6↓, MΦ apoptosis↓, bacterial clearance↑, splenocyte phagocytosis↑ | [41] | |||
| CLP | innate immune cells↑, MΦ↑, neutrophils↑ | [88] | |||
| Long-term survival following CLP | B cells↑, innate immune cells↑, MΦ↑ | [87] | |||
| MS-275 (entinostat) | HDAC1,2,3 | CLP | not improved | [41] | |
| LPS-dependent AKI | [95] | ||||
| KBH-A42 | pan | LPS-endotoxemia | TNF-α↓, IL-1β↓, IL-6↓, iNOS↓ | [96] | |
| Cyclic peptides | Romidepsin | pan | comorbidity sepsis | [97] | |
| Short chain fatty acids | Valproic acid | pan | LPS-dependent AKI | histological scores↓, MPO↓, NF-κB p65↓, NO↓, iNOS↓, TNF-α↓, IL-1β↓, nuclear HDAC3↓, cytosolic HDAC3↑ | [83] |
| Butyric acid | pan | CLP | ALI↓, neutrophil infiltra-tion↓, ICAM-1↓, E-selec- tin↓, IL-6 ↓, survival↑ | [80] | |
| CLP | long-term cognitive impairment↓ | [84] | |||
| SIRT-specific | EX-527 | SIRT1 | CLP | survival↑, TNF-α↓, IL-6↓, coagulopathy↓, bone marrow atrophy↓ | [69] |
| AGK2 | SIRT2 | CLP | survival↑, TNF-α↓, IL-6↓, clot formation↓, platelet function↓, bone marrow atrophy↓ | [91] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Knethen, A.; Brüne, B. Histone Deacetylation Inhibitors as Therapy Concept in Sepsis. Int. J. Mol. Sci. 2019, 20, 346. https://doi.org/10.3390/ijms20020346
von Knethen A, Brüne B. Histone Deacetylation Inhibitors as Therapy Concept in Sepsis. International Journal of Molecular Sciences. 2019; 20(2):346. https://doi.org/10.3390/ijms20020346
Chicago/Turabian Stylevon Knethen, Andreas, and Bernhard Brüne. 2019. "Histone Deacetylation Inhibitors as Therapy Concept in Sepsis" International Journal of Molecular Sciences 20, no. 2: 346. https://doi.org/10.3390/ijms20020346
APA Stylevon Knethen, A., & Brüne, B. (2019). Histone Deacetylation Inhibitors as Therapy Concept in Sepsis. International Journal of Molecular Sciences, 20(2), 346. https://doi.org/10.3390/ijms20020346