A Hybrid Hamiltonian for the Accelerated Sampling along Experimental Restraints


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Influence of the Renormalization Parameters on the Time-Dependent Relaxation Behavior in Simulations along Multiple Biasing Increments
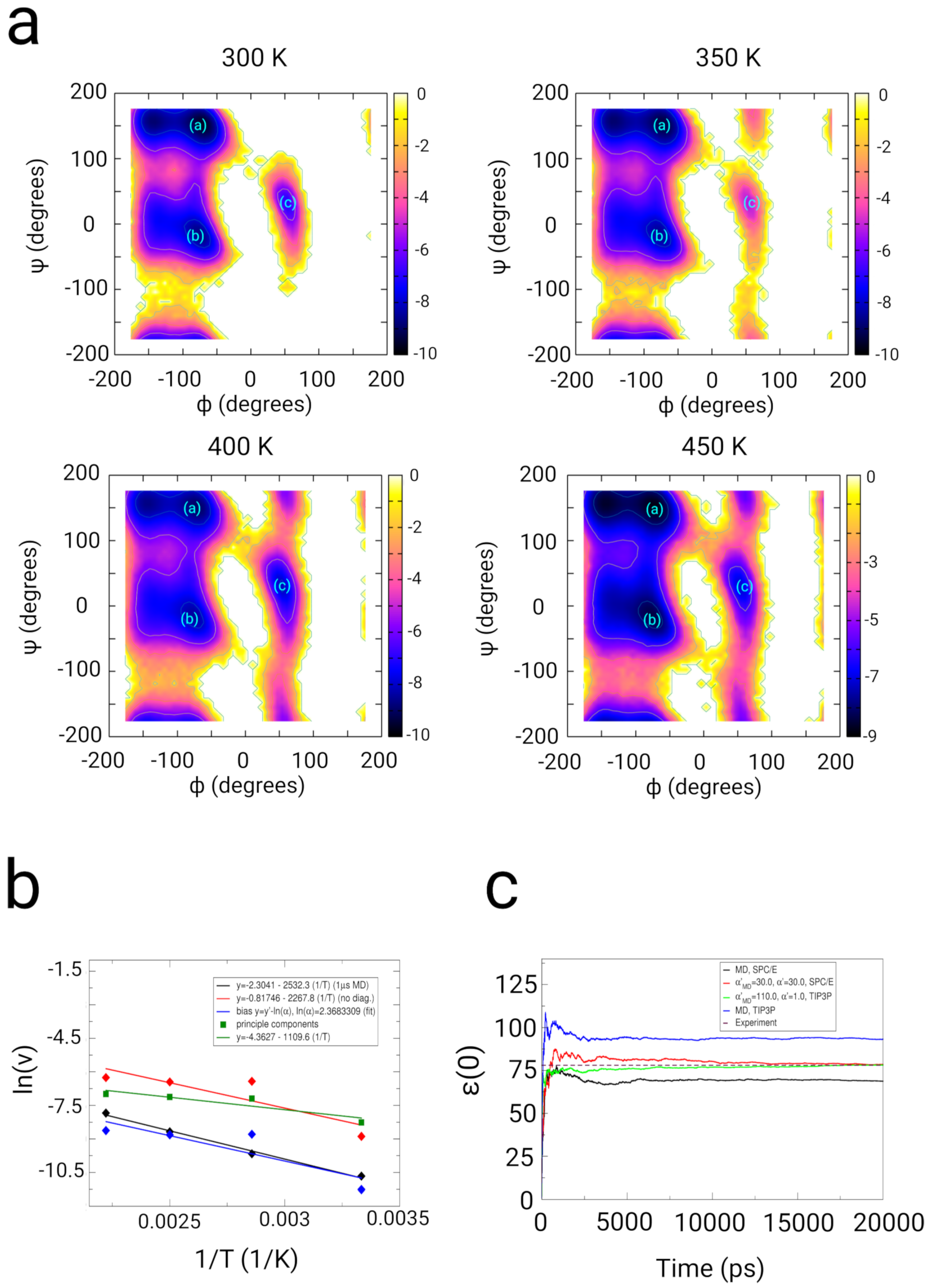
2.2. Dialanine
2.2.1. Results
2.2.2. Discussion
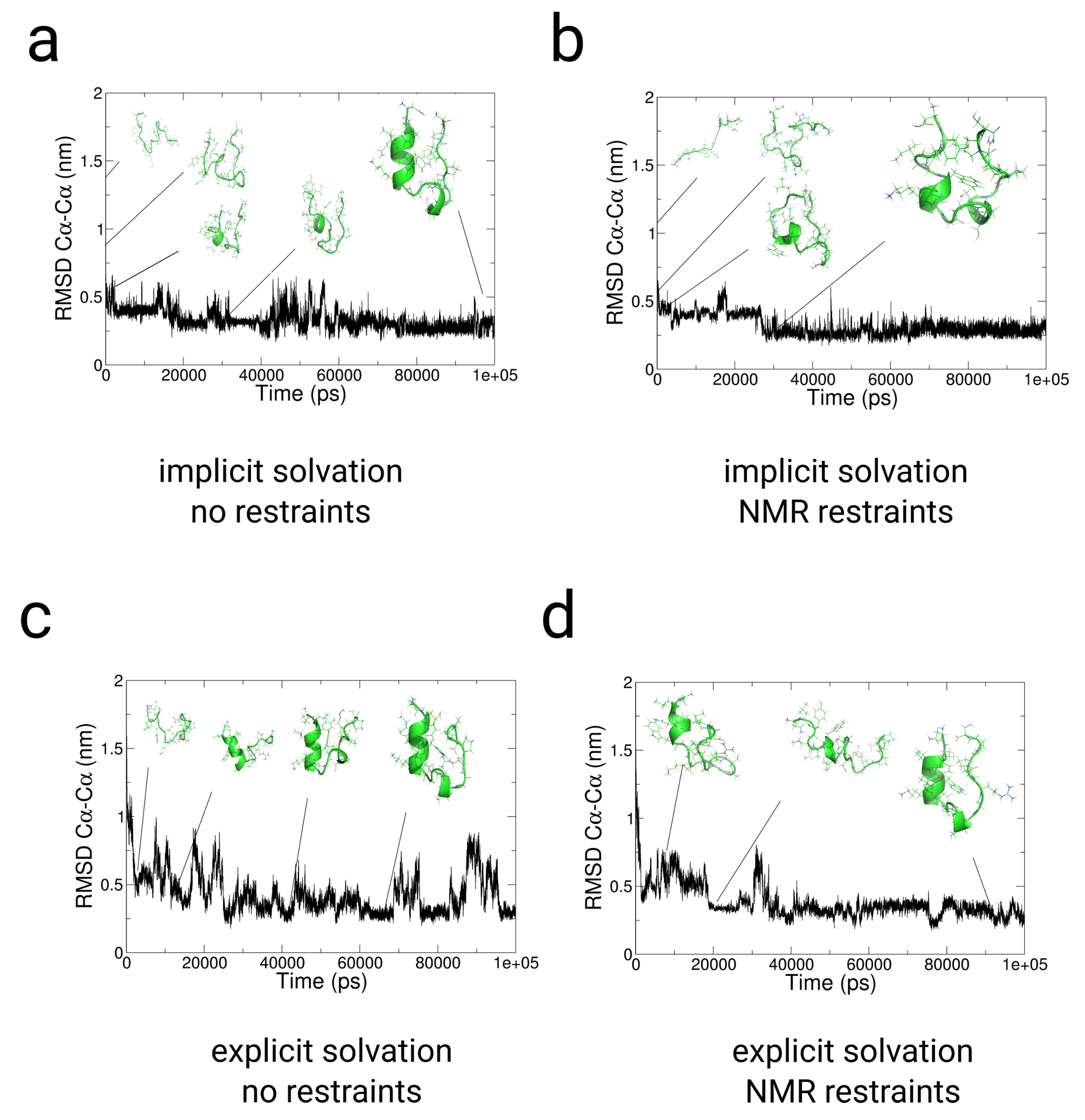
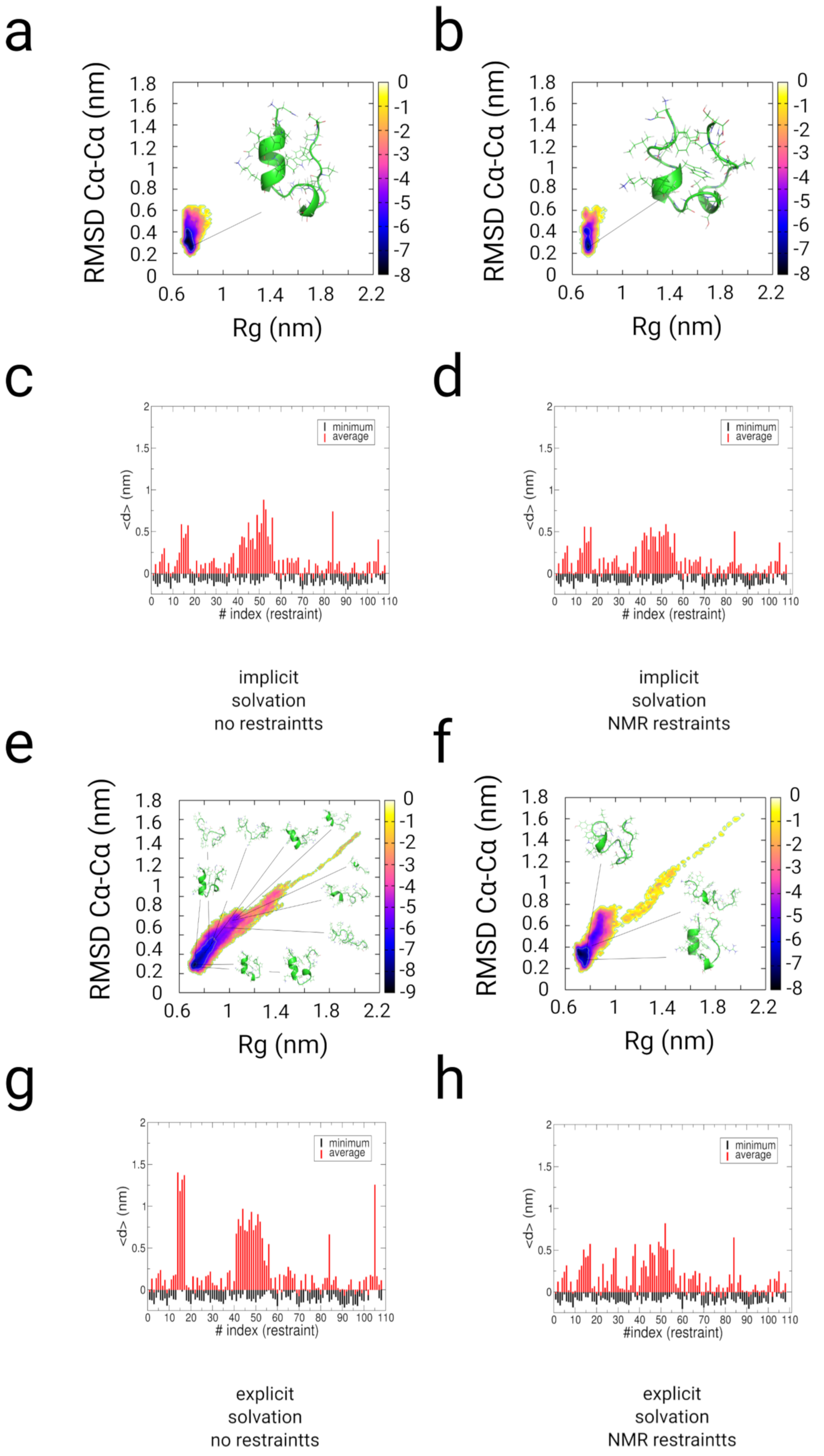
2.3. Simulations with Restraints from NMR NOESY spectra: TrpCage Folding
2.3.1. Results
2.3.2. Discussion
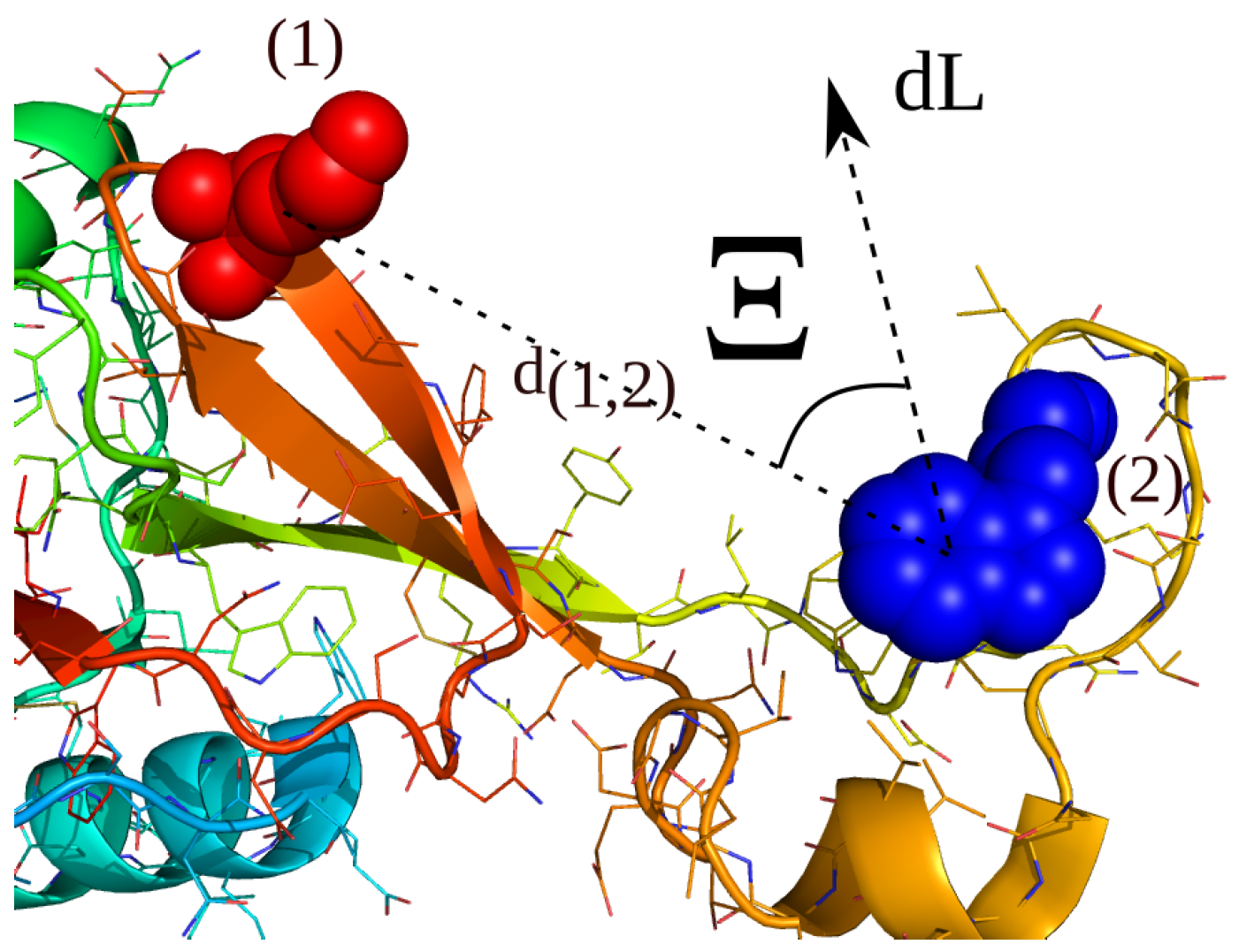
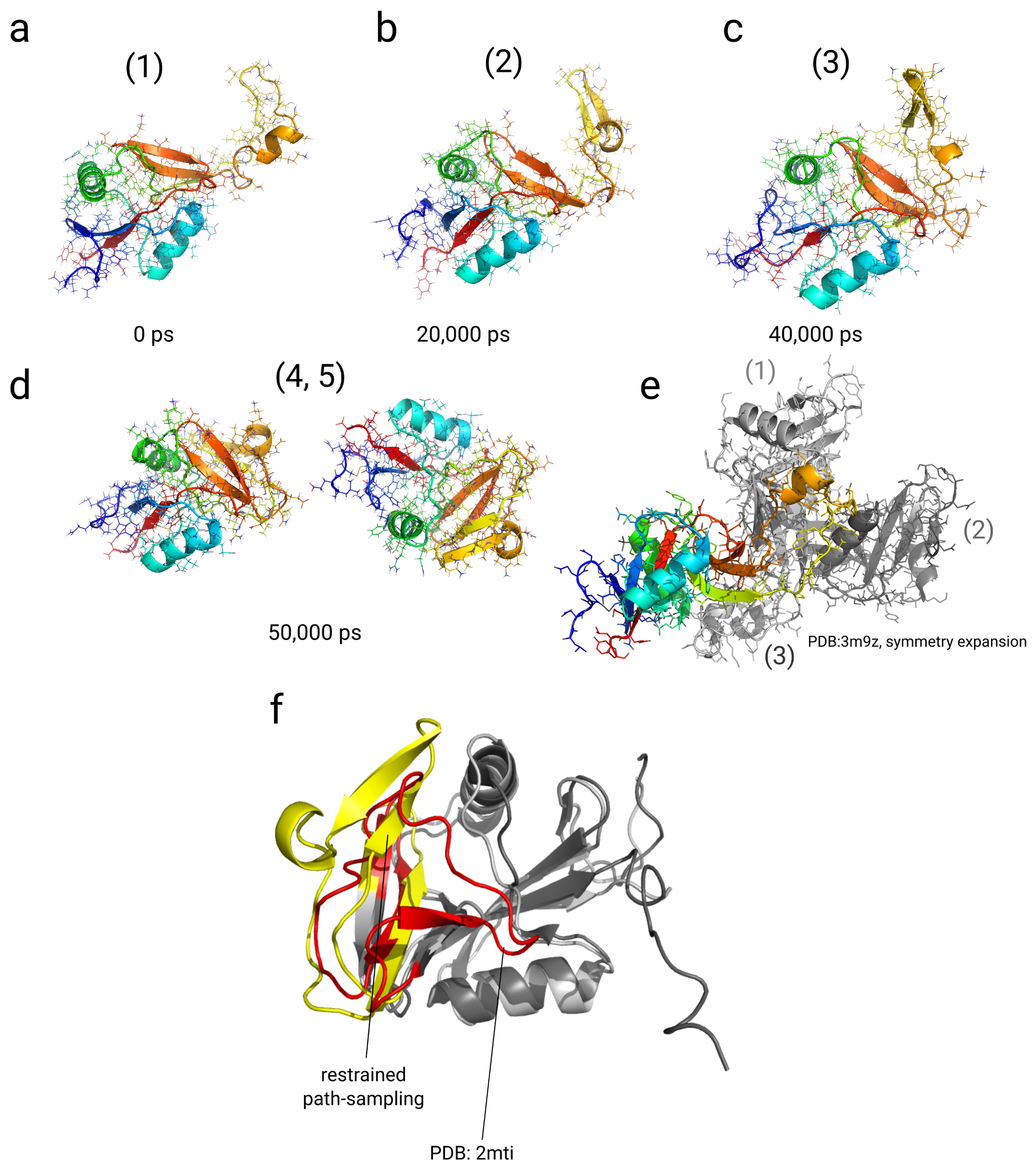
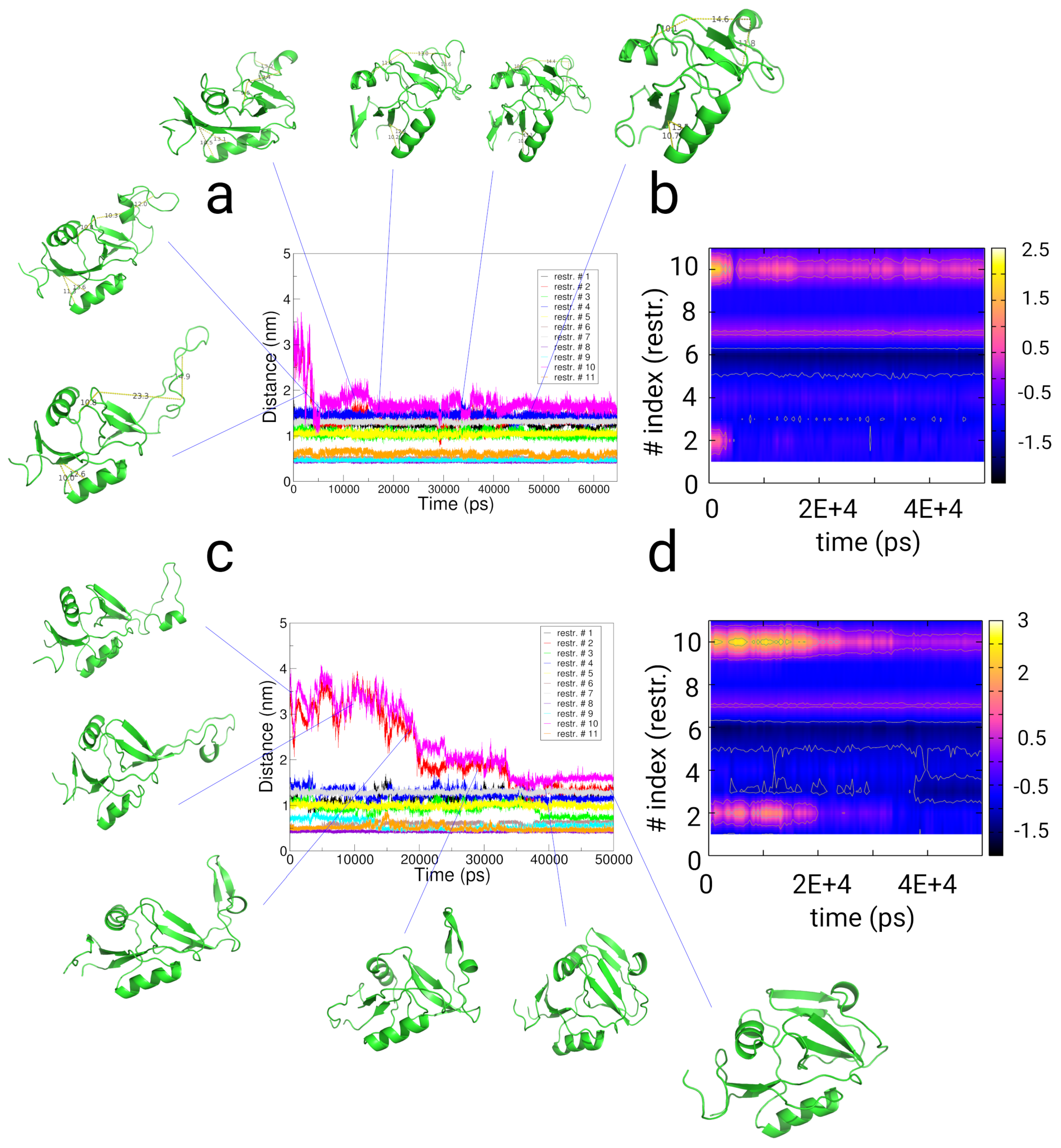
2.4. Simulations with Restraints from Chemical Crosslinking Data: NKR-P1A
2.4.1. Results
2.4.2. Discussion
3. Methods
3.1. Theory
Adaptive Restraint Modeling
3.2. Hybrid Hamiltonian
3.3. Restrained Hybrid Hamiltonian
- The momentum conservation is still obeyed and no additional energy is applied on the system, since the absolute value of varies from 0 to , due to for all possible values of .
- The effective biasing increments (, adaptive bias MD) and (, path-sampling) are modified for the atoms on which the restraint r is applied. That means that the path-increments are changed to and using a flexible bias vector in dependency of the vectorial quantity of and the restraint r, while the absolute increments applied per unit-time remain identical: .
3.4. Restrained Partitions
3.5. Path-Sampling Bias:
3.5.1. Theory
3.5.2. Adaptive Bias MD and Path-Sampling
3.6. Bias Gradients
- Start loop over MD steps.
- Loop over biases, N atoms.
- Determine the path-dependent increments at time t for the adaptive bias MD segment at periods of to define the biasing element (see Equation (28)).
- Evaluate to increase the history dependent bias at periods of for the bias with index i, atom k for the definition of (path-sampling) (see Equation (30)).
- Calculate gradients after renormalization in bias with index i, atom k (path-sampling and adaptive bias MD) (see Equation (7)).
- Determine the principal components of the 2 segments and .
- Evaluate overlap of bias with the given restraint to obtain (see Equation (12)).
- Add all gradients.
- , number of parallel biases
- , time-period for the adaptive bias MD for periods along parallel biases i for atoms with index k.
- , time-period for the path-sampling for periods along parallel biases i for atoms with index k.
- , fluctuation-width for the adaptive path-sampling algorithm to determine the deviation of the fluctuating bias around an average value . We used a constant value
- W, height of the Gaussians applied in the path-sampling. We applied a constant value kJ/mol ( kJ/mol).
- and , renormalization parameters of the bias to the unbiased gradient (see the definitions of and ).
- , fluctuation range of the unbiased gradient (see the definitions of and ).
- , fluctuation range of the bias related to the unbiased gradient.
- The list of atom-indices per residue are added to a list with a corresponding equilibrium distance . For the simulations of chemical crosslinking, we added all atom-indices for the first residue and one atom-index for the second residue.
3.7. Parameter Selection
3.8. Program and System Preparation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weber, G. Ligand binding and internal equilibiums in proteins. Biochemistry 1972, 11, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L. How Hofmeister ion interactions affect protein stability. Biophys. J. 1996, 71, 2056–2063. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Billeter, M.; Wagner, G.; Wüthrich, K. Solution NMR structure determination of proteins revisited. J. Biomol. NMR 2008, 42, 155–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.W.; Prlić, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Westbrook, J.D.; Woo, J.; et al. The RCSB Protein Data Bank: Views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, D345–D356. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Tildesley, D. Computer Simulation of Liquids; Clarendon Pr: Oxford, UK, 1987. [Google Scholar]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Cheatham, T.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.; Brooks, C.; MacKerell, A.; Nilsson, L.; Petrella, R.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comer, J.; Gumbart, J.C.; Hénin, J.; Lelièvre, T.; Pohorille, A.; Chipot, C. The adaptive biasing force method: Everything you always wanted to know but were afraid to ask. J. Phys. Chem. B 2015, 119, 1129–1151. [Google Scholar] [CrossRef]
- Shea, J.E.; Onuchic, J.; Brooks, C. Exploring the origins of topological frustration: Design of a minimally frustrated model of fragment B of protein A. Proc. Natl. Acad. Sci. USA 1999, 96, 12512–12517. [Google Scholar] [CrossRef] [Green Version]
- Shea, J.E.; Brooks, C.L., III. From folding theories to folding proteins: A review and assessment of simulation studies of protein folding and unfolding. Annu. Phys. Chem. Rev. 2001, 52, 499–535. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaendtner, J.; Bonomi, M. Efficient Sampling of High-Dimensional Free-Energy Landscapes with Parallel Bias Metadynamics. J. Chem. Theory Comput. 2015, 11, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Smiatek, J.; Heuer, A. Calculation of free energy landscapes: A histogram reweighted metadynamics approach. J. Comput. Chem. 2011, 32, 2084–2096. [Google Scholar] [CrossRef] [Green Version]
- Giberti, F.; Savalaglio, M.; Parrinello, M. Metadynamics studies of crystal nucleation. IUCr 2015, 2, 256–266. [Google Scholar] [CrossRef] [Green Version]
- Perego, C.; Savalaglio, M.; Parrinello, M. Molecular dynamics simulations of solutions at constant chemical potential. J. Chem. Phys. 2015, 142, 144113. [Google Scholar] [CrossRef] [Green Version]
- Schug, A.; Wenzel, W.; Hansmann, U.H.E. Energy landscape paving simulations of the trp-cage protein. J. Chem. Phys. 2005, 122, 194711. [Google Scholar] [CrossRef]
- Schug, A.; Herges, T.; Wenzel, W. Reproducible protein folding with the stochastic tunneling method. Phys. Rev. Lett. 2003, 91, 158102. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.R.; Voter, A.F. Temperature-accelerated dynamics for simulation of infrequent events. J. Chem. Phys. 2000, 112, 9599. [Google Scholar] [CrossRef]
- Montalenti, F.; Voter, A.F. Exploiting past visits or minimum-barrier knowledge to gain further boost in the temperature-accelerated dynamics method. J. Chem. Phys. 2002, 116, 4819. [Google Scholar] [CrossRef]
- Huber, T.; Torda, A.E.; van Gunsteren, W.F. Local elevation: A method for improving the searching properties of molecular dynamics simulation. J. Comput. Aided Mol. Des. 1994, 8, 695–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Brooks, C.L., III. λ-dynamics: A new approach to free energy calculations. J. Chem. Phys. 1996, 105, 2414. [Google Scholar] [CrossRef]
- Knight, J.L.; Brooks, C.L., III. Multi-Site λ-dynamics for simulated Structure-Activity Relationship studies. J. Chem. Theor. Comput. 2011, 7, 2728–2739. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange multicanonical algorithm and multicanonical replica-exchange method for simulating systems with rough energy landscape. Chem. Phys. Lett. 2000, 329, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Replica-exchange multicanonical and multicanonical replica-exchange Monte Carlo simulations of peptides. I. Formulation and benchmark test. J. Chem. Phys. 2003, 118, 6664. [Google Scholar] [CrossRef]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Replica-exchange multicanonical and multicanonical replica-exchange Monte Carlo simulations of peptides. II. Application to a more complex system. J. Chem. Phys. 2003, 118, 6676. [Google Scholar] [CrossRef]
- Fukunishi, H.; Watanabe, O.; Takada, S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J. Chem. Phys. 2002, 116, 9058. [Google Scholar] [CrossRef]
- Faller, R.; Yan, Q.; de Pablo, J.J. Multicanonical parallel tempering. J. Chem. Phys. 2002, 116, 5419. [Google Scholar] [CrossRef]
- Whitfield, T.W.; Bu, L.; Straub, J.E. Generalized parallel sampling. Phys. A 2002, 305, 157–171. [Google Scholar] [CrossRef]
- Jang, S.; Shin, S.; Pak, Y. Replica-exchange method using the generalized effective potential. Phys. Rev. Lett. 2003, 91, 058305. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Huang, X.; Zhou, R.; Berne, B.J. Hydrophobic aided replica exchange: An efficient algorithm for protein folding in explicit solvent. J. Phys. Chem. B 2006, 110, 19018–19022. [Google Scholar] [CrossRef]
- Cheng, X.; Cui, G.; Hornak, V.; Simmerling, C. Modified replica exchange simulation methods for local structure refinement. J. Phys. Chem. B 2005, 109, 8220–8230. [Google Scholar] [CrossRef]
- Lyman, E.; Ytreberg, M.; Zuckerman, D.M. Resolution exchange simulation. Phys. Rev. Lett. 2006, 96, 028105. [Google Scholar] [CrossRef]
- Liu, P.; Voth, G.A. Smart resolution replica exchange: An efficient algorithm for exploring complex energy landscapes. J. Chem. Phys. 2007, 126, 045106. [Google Scholar] [CrossRef]
- Calvo, F. All-exchanges parallel tempering. J. Chem. Phys. 2005, 123, 124106. [Google Scholar] [CrossRef]
- Rick, S.W. Replica exchange with dynamical scaling. J. Chem. Phys. 2007, 126, 054102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamberaj, H.; van der Vaart, A. Multiple scaling replica exchange for the conformational sampling of biomolecules in explicit water. J. Chem. Phys. 2007, 127, 234102. [Google Scholar] [CrossRef] [PubMed]
- Brenner, P.; Sweet, C.R.; VonHandorf, D.; Izaguirre, J.A. Accelerating the replica exchange method through an efficient all-pairs exchange. J. Chem. Phys. 2007, 126, 074103. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, J. Simulation via direct computation of partition functions. Phys. Rev. E 2007, 76, 036708. [Google Scholar] [CrossRef]
- Trebst, S.; Troyer, M.; Hansmann, U.H.E. Optimized parallel tempering simulations of proteins. J. Chem. Phys. 2006, 124, 174903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolhuis, P.G.; Dellago, C.; Chandler, D. Reaction coordinates of biomolecular isomerization. Proc. Natl. Acad. Sci. USA 2000, 97, 5877. [Google Scholar] [CrossRef] [PubMed]
- Bello-Rivas, J.M.; Elber, R. Exact milestoning. J. Chem. Phys. 2015, 142, 094102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, A.J.; Jarzynski, C. Replica exchange with nonequilibrium switches. Proc. Natl. Acad. Sci. USA 2009, 106, 12224–12229. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.W.; Jasnow, D.; Zuckerman, D.M. The “weighted ensemble” path sampling method is statistically exact for a broad class of stochastic processes and binning procedures. J. Chem. Phys. 2010, 132, 054107. [Google Scholar] [CrossRef] [Green Version]
- a Beccara, S.; Škripić, T.; Covino, R.; Faccioli, P. Dominant folding pathways of a WW domain. Proc. Natl. Acad. Sci. USA 2012, 109, 2330–2335. [Google Scholar] [CrossRef] [Green Version]
- a Beccara, S.; Fant, L.; Faccioli, P. Variational scheme to compute protein reaction pathways using atomistic force fields with explicit solvent. Phys. Rev. Lett. 2015, 114, 098103. [Google Scholar] [CrossRef] [PubMed]
- Elber, R. Computer simulations of long time dynamics. J. Chem. Phys. 2016, 144, 060901. [Google Scholar] [CrossRef] [PubMed]
- Olender, R.; Elber, R. Calculation of classical trajectories with a very large time step: Formalism and numerical examples. J. Chem. Phys. 1996, 105, 9299–9315. [Google Scholar] [CrossRef]
- Ma, Q.; Izaguirre, J.A. Targeted Mollified Impulse: A Multiscale Stochastic Integrator for Long Molecular Dynamics Simulations. Multiscale Model. Simul. 2003, 2, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Leimkuhler, B.; Margul, D.T.; Tuckerman, M.E. Molecular dynamics based enhanced sampling of collective variables with very large time steps. Mol. Phys. 2013, 111, 3579–3594. [Google Scholar] [CrossRef]
- Richters, D.; Kühne, T.D. Linear-scaling self-consistent field theory based molecular dynamics: Application to C60buckyballs colliding with graphite. Mol. Sim. 2018, 44, 1380–1386. [Google Scholar] [CrossRef]
- Chen, W.; Ferguson, A.L. Molecular enhanced sampling with autoencoders: On-the-fly collective variable discovery and accelerated free energy landscape exploration. J. Comput. Chem. 2018, 39, 2079–2102. [Google Scholar] [CrossRef] [PubMed]
- Chiavazzo, E.; Covino, R.; Coifman, R.R.; Gear, C.W.; Georgiou, A.S.; Hummer, G.; Kevredikis, I.G. Intrinsic map dynamics exploration for uncharted effective free-energy landscapes. Proc. Natl. Acad. Sci. USA 2017, 114, E5494–E5503. [Google Scholar] [CrossRef]
- Chen, M.; Yu, T.Q.; Tuckerman, M.E. Locating landmarks on high-dimensional free energy surfaces. Proc. Natl. Acad. Sci. USA 2015, 112, 3235–3240. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Doyle, J.P.K. Entropic tempering: A method for overcoming quasiergodicity in simulation. Phys. Rev. E 2000, 63, 010902. [Google Scholar] [CrossRef]
- Morris-Andrews, A.; Rottler, J.; Plotkin, S.S. A systematically coarse-grained model for DNA and its predictions for persistence length, stacking, twist, and chirality. J. Chem. Phys. 2010, 132, 035105. [Google Scholar] [CrossRef] [Green Version]
- Ouldridge, T.E.; Louis, A.A.; Doyle, J.P.K. Structural, mechanical, and thermodynamic properties of a coarse-grained DNA model. J. Chem. Phys. 2011, 134, 085101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naôme, A.; Laaksonen, A.; Vercauteren, D.P. A Solvent-Mediated Coarse-Grained Model of DNA Derived with the Systematic Newton Inversion Method. J. Chem. Theor. Comput. 2014, 10, 3541–3549. [Google Scholar] [CrossRef]
- Takada, S. Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct. Biol. 2012, 22, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Morris-Andrews, A.; Brown, F.L.; Shea, J.E. A Coarse-Grained Model for Peptide Aggregation on a Membrane Surface. J. Phys. Chem. B 2014, 118, 8420–8432. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theor. Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [Green Version]
- Schwieters, C.D.; Bermejo, G.A.; Clore, G.M. Xplor-NIH for molecular structure determination from NMR and other data sources. Prot. Sci. 2018, 27, 26–40. [Google Scholar] [CrossRef]
- Clore, G.M.; Gronenborn, A.M.; Kjaer, M.; Poulsen, F.M. The determination of the three-dimensional structure of barley serine proteinase inhibitor 2 by nuclear magnetic resonance, distance geometry and restrained molecular dynamics. Prot. Eng. 1987, 1, 305–311. [Google Scholar] [CrossRef]
- Clore, G.M.; Nilges, M.; Sukumaran, D.K.; Brünger, A.T.; Karplus, M.; Gronenborn, A.M. The three-dimensional structure of alpha1-purothionin in solution: Combined use of nuclear magnetic resonance, distance geometry and restrained molecular dynamics. EMBO J. 1986, 5, 2729–2735. [Google Scholar] [CrossRef]
- Robustelli, P.; Kohlhoff, K.; Cavalli, A.; Vendruscolo, M. Using NMR chemical shifts as structural restraints in molecular dynamics simulations of proteins. Structure 2010, 18, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bonomi, M.; Parrinello, M. Linking well-tempered metadynamics simulations with experiments. Biophys. J. 2010, 98, L44–L46. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Han, W.; Fiorin, G.; Islam, S.M.; Schulten, K.; Roux, B. Structural Refinement of Proteins by Restrained Molecular Dynamics Simulations with Non-interacting Molecular Fragments. PLoS Comput. Biol. 2015, 11, e1004368. [Google Scholar] [CrossRef]
- Islam, S.M.; Roux, B. Simulating the distance distribution between spin-labels attached to proteins. J. Phys. Chem. B 2015, 119, 3901–3911. [Google Scholar] [CrossRef]
- Granata, D.; Camilloni, C.; Vendruscolo, M.; Laio, A. Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 6817–6822. [Google Scholar] [CrossRef]
- Ma, T.; Zang, T.; Wang, Q.; Ma, J. Refining protein structures using enhanced sampling techniques with restraints derived from an ensemble-based model. Prot. Sci. 2018, 27, 1842–1849. [Google Scholar] [CrossRef]
- Chen, P.-c.; Hub, J.S. Validating solution ensembles from molecular dynamics simulation by wide-angle X-ray scattering data. Biophys. J. 2014, 107, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S. Interpreting solution X-ray scattering data using molecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 18–26. [Google Scholar] [CrossRef]
- Björling, A.; Niebling, S.; Marcellini, M.; van der Spoel, D.; Westenhoff, S. Deciphering solution scattering data with experimentally guided molecular dynamics simulations. J. Chem. Theor. Comput. 2015, 11, 780–787. [Google Scholar] [CrossRef]
- Velásquez-Muriel, J.; Lasker, K.; Russel, D.; Phillips, J.; Webb, B.M.; Schneidmann-Duhovny, D.; Sali, A. Assembly of macromolecular complexes by satisfaction of spatial restraints from electron microscopy images. Proc. Natl. Acad. Sci. USA 2012, 109, 18821–18826. [Google Scholar] [CrossRef]
- Park, H.; DiMaio, F.; Baker, D. The origin of consistent protein structure refinement from structural averaging. Structure 2015, 23, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, U.; Ulyanov, N.B.; Kumar, A.; James, T.L. Molecular dynamics with weighted time-averaged restraints for a DNA octamer. Dynamic interpretation of nuclear magnetic resonance data. J. Mol. Biol. 1993, 234, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Scheek, R.M.; Torda, A.E.; Kemmink, J.; van Gunsteren, W.F. Computational Aspects of the Study of Biological Macromolecules by Nuclear Magnetic Resonance Spectroscopy; Plenum Press: New York, NY, USA, 1991; pp. 209–217. [Google Scholar]
- Fennen, J.; Torda, A.E.; van Gunsteren, W.F. Structure refinement with molecular dynamics and a Boltzmann-weighted ensemble. J. Biomol. NMR 1995, 6, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Hansen, N.; Heller, F.; Schmid, N.; van Gunsteren, W.F. Time-averaged order parameter restraints in molecular dynamics simulations. J. Biomol. NMR 2014, 60, 169–187. [Google Scholar] [CrossRef]
- Merkley, E.D.; Rysavy, S.; Kahraman, A.; Hafen, R.P.; Daggett, V.; Adkins, J.N. Distance restraints from crosslinking mass spectrometry: Mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Prot. Sci. 2013, 23, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Rozbeský, D.; Man, P.; Kavan, D.; Chmelík, J.; Černý, J.; Bezouška, K.; Novák, P. Chemical cross-linking and H/D exchange for fast refinement of protein crystal structure. Anal. Chem. 2012, 84, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Peter, E.K.; Shea, J.E. An adaptive bias-hybrid MD/kMC algorithm for protein folding and aggregation. Phys. Chem. Chem. Phys. 2017, 19, 17373–17382. [Google Scholar] [CrossRef]
- Peter, E.K.; Černý, J. Enriched Conformational Sampling of DNA and Proteins with a Hybrid Hamiltonian Derived from the Protein Data Bank. Int. J. Mol. Sci. 2018, 19, 3405. [Google Scholar] [CrossRef]
- Peter, E.K. Adaptive enhanced sampling with a path-variable for the simulation of protein folding and aggregation. J. Chem. Phys. 2017, 147, 214902. [Google Scholar] [CrossRef]
- Neidigh, J.W.; Fesinmeyer, R.M. Designing a 20-residue protein. Nat. Struct. Biol. 2002, 9, 425–430. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation—Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Wang, H.; Junghans, C.; Kremer, K. Comparative atomistic and coarse-grained study of water: What do we lose by coarse-graining? Eur. Phys. J. E 2009, 28, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izvekov, S.; Parrinello, M.; Burnham, C.J.; Voth, G.A. Effective force fields for condensed phase systems from ab initio molecular dynamics simulation: A new method for force-matching. J. Chem. Phys. 2004, 120, 10896–10913. [Google Scholar] [CrossRef] [PubMed]
- Soper, A.K.; Phillips, M.G. A new determination of the structure of water at 25 °C. Chem. Phys. 1986, 107, 47. [Google Scholar] [CrossRef]
- Mills, R. Self-diffusion in normal and heavy water in the range 1–45.deg. J. Phys. Chem. 1973, 77, 685. [Google Scholar] [CrossRef]
- Price, W.S.; Ide, H.; Arata, Y. Self-Diffusion of Supercooled Water to 238 K Using PGSE NMR Diffusion Measurements. J. Phys. Chem. A 1999, 103, 448. [Google Scholar] [CrossRef]
- Owen, B.B.; Miller, R.C.; Milner, C.E.; Cogan, H.L. The dielectric constant of water as a function of temperature and pressure. J. Phys. Chem. 1961, 65, 2065–2070. [Google Scholar] [CrossRef]
- Braun, D.; Boresch, S.; Steinhauser, O. Transport and dielectric properties of water and the influence of coarse-graining: Comparing BMW, SPC/E, and TIP3P models. J. Chem. Phys. 2014, 140, 064107. [Google Scholar] [CrossRef]
- Tobias, D.J.; Brooks, C.L., III. Conformational equilibrium in the alanine dipeptide in the gas phase and aqueous solution: A comparison of theoretical results. J. Phys. Chem. 1992, 96, 3864–3870. [Google Scholar] [CrossRef]
- Swope, W.C.; Pitera, J.W.; Suits, F.; Pitman, M.; Eleftheriou, M.; Fitch, B.G.; Germain, R.S.; Rayshubski, A.; Zhestkov, Y.; Zhou, R. Describing Protein Folding Kinetics by Molecular Dynamics Simulations. 2. Example Applications to Alanine Dipeptide and a β-Hairpin Peptide. J. Chem. Phys. B 2004, 108, 6582–6594. [Google Scholar] [CrossRef]
- Stelzl, L.S.; Hummer, G. Kinetics from Replica Exchange Molecular Dynamics Simulations. J. Chem. Theor. Comput. 2017, 13, 3927–3935. [Google Scholar] [CrossRef]
- Tiwary, P.; Parrinello, M. From metadynamics to dynamics. Phys. Rev. Lett. 2013, 111, 230602. [Google Scholar] [CrossRef]
- Peter, E.K.; Shea, J.E. A hybrid MD-kMC algorithm for folding proteins in explicit solvent. Phys. Chem. Chem. Phys. 2014, 16, 6430–6440. [Google Scholar] [CrossRef] [PubMed]
- Peter, E.K.; Pivkin, I.V.; Shea, J.E. A kMC-MD method with generalized move-sets for the simulation of folding of α-helical and β-stranded peptides. J. Chem. Phys. 2015, 142, 144903. [Google Scholar] [CrossRef] [PubMed]
- Culik, R.M.; Serrano, A.L.; Bunagan, M.R.; Gai, F. Achieving secondary structural resolution in kinetic measurements of protein folding: A case study of the folding mechanism of Trp-cage. Angew. Chem. 2011, 123, 11076–11079. [Google Scholar] [CrossRef]
- Meuzelaar, H.; Marino, K.A.; Huerta-Viga, A.; Panman, M.R.; Smeenk, L.E.J.; Kettelarij, A.J.; van Maarseveen, P.T.J.H.; Bolhuis, P.G.; Woutersen, S. Folding Dynamics of the Trp-Cage Miniprotein: Evidence for a Native-Like Intermediate from Combined Time-Resolved Vibrational Spectroscopy and Molecular Dynamics Simulations. J. Phys. Chem. B 2013, 117, 11490–11501. [Google Scholar] [CrossRef]
- Juraszek, J.; Bolhuis, P.G. Sampling the multiple folding mechanisms of Trp-cage in explicit solvent. Proc. Natl. Acad. Sci. USA 2006, 103, 15859–15864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juraszek, J.; Bolhuis, P.G. Rate constant and reaction coordinate of Trp-cage folding in explicit water. Biophys. J. 2008, 95, 4246–4257. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, F.; Pietrucci, F.; Laio, A.; Piana, S. A Kinetic Model of Trp-Cage Folding from Multiple Biased Molecular Dynamics Simulations. PLoS Comput. Biol. 2009, 5, e1000452. [Google Scholar] [CrossRef]
- Snow, C.D.; Zagrovic, B.; Pande, V.S. The Trp Cage: Folding Kinetics and Unfolded State Topology via Molecular Dynamics Simulations. J. Am. Chem. Soc. 2002, 124, 14548–14549. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Lai, Z.; Biggs, J.D.; Wang, J.; Mukamel, S. Two-dimensional stimulated resonance Raman spectroscopy study of the Trp-cage peptide folding. Phys. Chem. Chem. Phys. 2013, 15, 19457–19464. [Google Scholar] [CrossRef] [PubMed]
- Neuweiler, H.; Doose, S.; Sauer, M. A microscopic view of miniprotein folding: Enhanced folding efficiency through formation of an intermediate. Proc. Natl. Acad. Sci. USA 2005, 102, 16650–16655. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Pabit, S.A.; Roitberg, A.E.; Hagen, S.J. Smaller and Faster: The 20-Residue Trp-Cage Protein Folds in 4 μs. J. Am. Chem. Soc. 2002, 124, 12952–12953. [Google Scholar] [CrossRef]
- Kolenko, P.; Rozbeský, D.; Vaněk, O.; Kopecký, V.; Hofbauerová, K.; Novák, P.; Pompach, P.; Hašek, J.; Skálová, T.; Bezouška, K.; et al. Molecular architecture of mouse activating NKR-P1 receptors. J. Struct. Biol. 2011, 175, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Rozbeský, D.; Adámek, D.; Pospíšilová, E.; Novák, P.; Chmelík, J. Solution structure of the lymphocyte receptor Nkrp1a reveals a distinct conformation of the long loop region as compared to in the crystal structure. Proteins 2016, 84, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Skálová, T.; Kotýnková, K.; Dušková, J.; Hašek, J.; Koval’, T.; Kolenko, P.; Novák, P.; Man, P.; Hanč, P.; Vaněk, O.; et al. Mouse Clr-g, a ligand for NK cell activation receptor NKR-P1F: Crystal structure and biophysical properties. J. Immunol. 2012, 189, 4881–4889. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, N.A.; Xi, W.; Wang, W.; Hansmann, U.H.E. Simulating Protein Fold Switching by Replica Exchange with Tunneling. J. Chem. Theory Comput. 2016, 12, 5656–5666. [Google Scholar] [CrossRef]
- Kleinert, H. Path Integrals in Quantum Mechanics, Statistics, Polymer Physics, and Financial Markets, 5th ed.; World Scientific: Singapore, 2009; pp. 1–1547. [Google Scholar]
- Feynman, R.; Hibbs, A.R. Quantum Mechanics and Path Integrals; MacGraw Hill Companies: New York, NY, USA, 1965. [Google Scholar]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef]
- Balsera, M.A.; Wriggers, W.; Ono, Y.; Schulten, K. Principal Component Analysis and Long Time Protein Dynamics. J. Phys. Chem. 1996, 100, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Kollman, P.A. Advances and Continuing Challenges in Achieving Realistic and Predictive Simulations of the Properties of Organic and Biological Molecules. Acc. Chem. Res. 1996, 29, 461–469. [Google Scholar] [CrossRef]
- Hess, B.; van der Spoel, D.; Lindahl, E. The Gromacs Development Team. 2012. Available online: www.gromacs.org (accessed on 12 January 2018).
- Grubmüller, H. Predicting slow structural transitions in macromolecular systems: Conformational flooding. Phys. Rev. E 1995, 52, 2893. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, E.K.; Černý, J. A Hybrid Hamiltonian for the Accelerated Sampling along Experimental Restraints. Int. J. Mol. Sci. 2019, 20, 370. https://doi.org/10.3390/ijms20020370
Peter EK, Černý J. A Hybrid Hamiltonian for the Accelerated Sampling along Experimental Restraints. International Journal of Molecular Sciences. 2019; 20(2):370. https://doi.org/10.3390/ijms20020370
Chicago/Turabian StylePeter, Emanuel K., and Jiří Černý. 2019. "A Hybrid Hamiltonian for the Accelerated Sampling along Experimental Restraints" International Journal of Molecular Sciences 20, no. 2: 370. https://doi.org/10.3390/ijms20020370
APA StylePeter, E. K., & Černý, J. (2019). A Hybrid Hamiltonian for the Accelerated Sampling along Experimental Restraints. International Journal of Molecular Sciences, 20(2), 370. https://doi.org/10.3390/ijms20020370