ADAR1p150 Forms a Complex with Dicer to Promote miRNA-222 Activity and Regulate PTEN Expression in CVB3-Induced Viral Myocarditis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
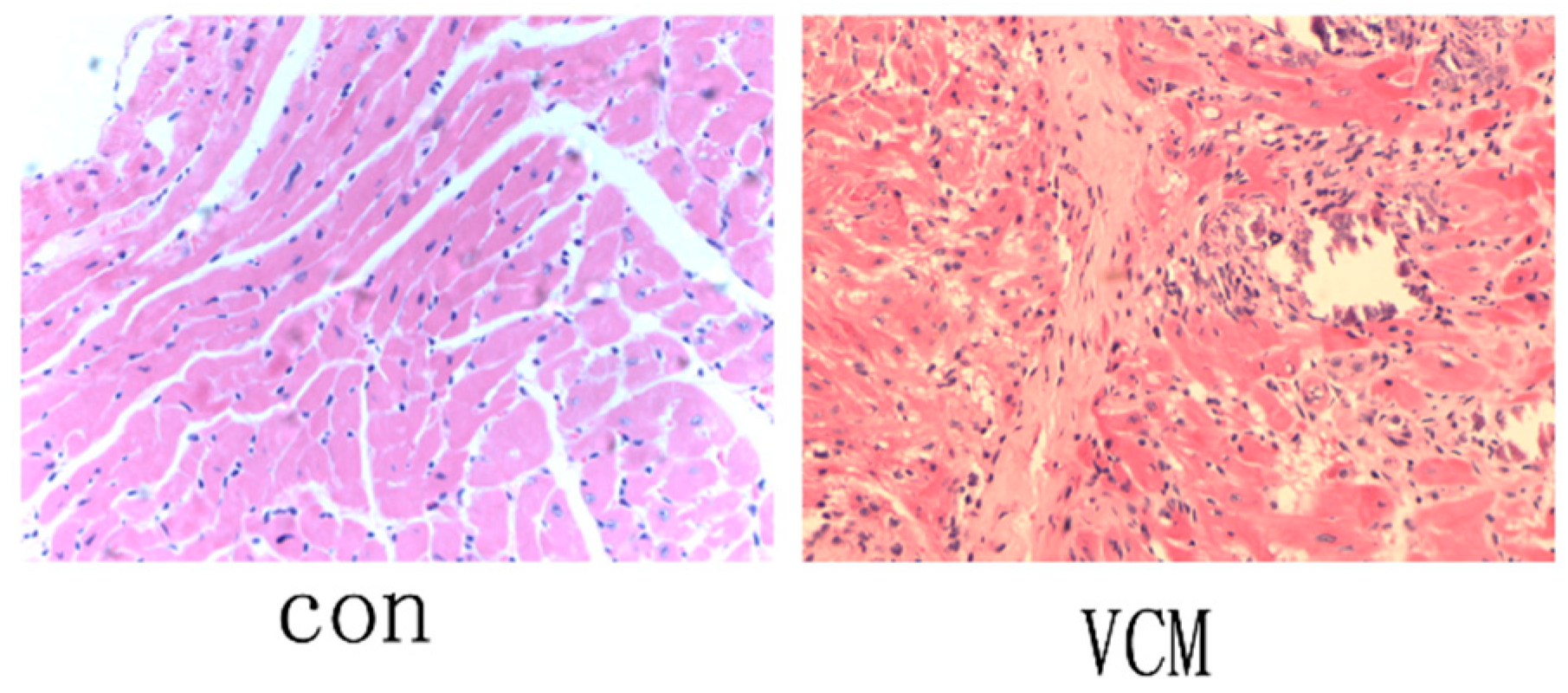
2.1. Verification of the VMC Mouse Model
2.2. ADAR1 Expression in the Mouse Model of VMC and Cardiac Cell Lines Infected with CBV3
2.3. Interaction between ADAR1 and Dicer in the Hearts fromVMC Mice and in CBV3-Infected H9c2 Cells
2.4. Increased Level of miRNA-222 in the Hearts of VMC Mice and in CBV3-Infected H9c2 Cells
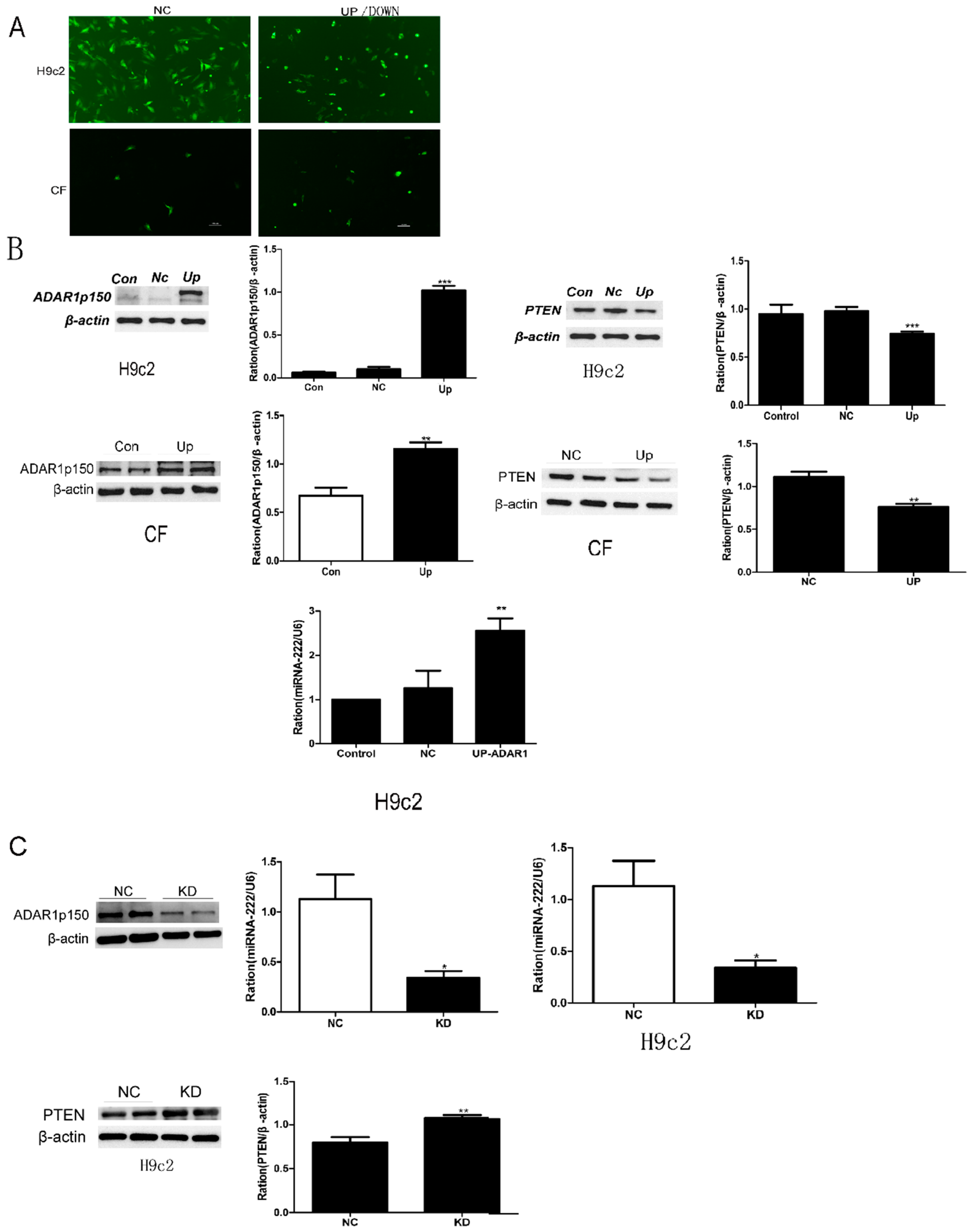
2.5. Effects of ADAR1p150 on miRNA-222 Synthesis in Cultured Cells
2.6. miR-222 Downregulation ofPTEN Expression
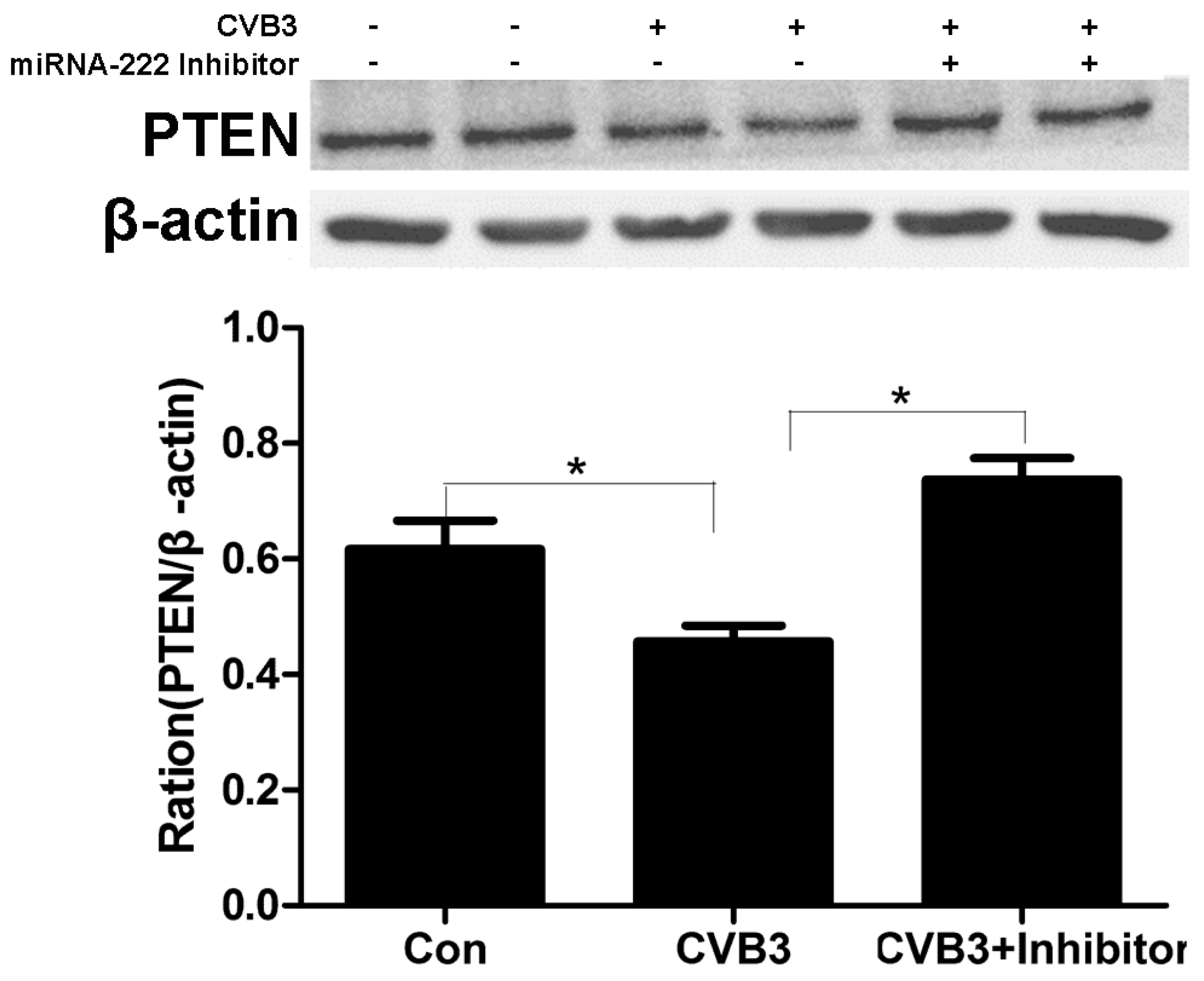
2.7. Regulation of PTEN by miRNA-222 Triggered by ADAR1p150 in CVB3-Induced Myocarditis
2.8. ADAR1p150 Plays an Important Role in Maintaining Cell Viability by Regulating PTEN Expression
3. Discussion
4. Materials and methods
4.1. Animals
4.2. VMC Mouse Model
4.3. Histology and Immunohistochemistry
4.4. Culture of Cardiomyocytes and Fibroblasts
4.5. H9c2 Cell Culture
4.6. Western Blots
4.7. Coimmunoprecipitation (Co-IP)
4.8. Upregulation and Knockdownof ADAR1p150 Protein
4.9. miRNA Synthesis and miRNA Inhibitor
4.10. Transfection of miRNAs
4.11. RNA Purification andReverse Transcription Polymerase Chain Reaction (RT-PCR)
4.12. Analysis of Cardiomyocyte Viability
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cooper, L.T. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Pankuweit, S.; Klingel, K. Viral myocarditis: From experimental models to molecular diagnosis in patients. Heart Fail. Rev. 2013, 18, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Liu, P.P.; Cooper, L.T. Myocarditis. Lancet Lond. Engl. 2012, 379, 738–747. [Google Scholar] [CrossRef]
- Esfandiarei, M.; McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol. 2008, 3, 127–155. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.E. Coxsackievirus myocarditis: Interplay between virus and host in the pathogenesis of heart disease. Viral Immunol. 2006, 19, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Savvatis, K.; Miteva, K.; Peng, J.; Ringe, J.; Warstat, K.; Schmidt-Lucke, C.; Sittinger, M.; Schultheiss, H.P.; Tschope, C. Mesenchymal stem cells improve murine acute coxsackievirus B3-induced myocarditis. Eur. Heart J. 2011, 32, 2168–2178. [Google Scholar] [CrossRef]
- Chow, L.H.; Beisel, K.W.; McManus, B.M. Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab. Investig. 1992, 66, 24–31. [Google Scholar]
- Yuan, J.P.; Zhao, W.; Wang, H.T.; Wu, K.Y.; Li, T.; Guo, X.K.; Tong, S.Q. Coxsackievirus B3-induced apoptosis and caspase-3. Cell Res. 2003, 13, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.W.; O’Connell, J.B.; Herskowitz, A.; Rose, N.R.; McManus, B.M.; Billingham, M.E.; Moon, T.E. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N. Engl. J. Med. 1995, 333, 269–275. [Google Scholar] [CrossRef]
- Wakafuji, S.; Okada, R. Twenty year autopsy statistics of myocarditis incidence in Japan. Jpn. Circ. J. 1986, 50, 1288–1293. [Google Scholar] [CrossRef]
- Gravanis, M.B.; Sternby, N.H. Incidence of myocarditis. A 10-year autopsy study from Malmo, Sweden. Arch. Pathol. Lab. Med. 1991, 115, 390–392. [Google Scholar] [PubMed]
- Carniel, E.; Sinagra, G.; Bussani, R.; Di Lenarda, A.; Pinamonti, B.; Lardieri, G.; Silvestri, F. Fatal myocarditis: Morphologic and clinical features. Ital. Heart J. 2004, 5, 702–706. [Google Scholar] [PubMed]
- Ward, C. Severe arrhythmias in coxsackievirus B3 myopericarditis. Arch. Dis. Child. 1978, 53, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Bendig, J.W.A.; O’Brien, P.S.; Muir, P.; Porter, H.J.; Caul, E.O. Enterovirus sequences resembling coxsackievirus A2 detected in stool and spleen from a girl with fatal myocarditis. J. Med. Virol. 2001, 64, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Keegan, L.P.; Leroy, A.; Sproul, D.; O’Connell, M.A. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 2004, 5, 209. [Google Scholar] [CrossRef]
- George, C.X.; Gan, Z.; Liu, Y.; Samuel, C.E. Adenosine deaminases acting on RNA (ADARs), RNA editing and interferon action. J. Interferon Cytokine Res. 2011, 31, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The ADAR protein family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heale, B.S.; Keegan, L.P.; McGurk, L.; Michlewski, G.; Brindle, J.; Stanton, C.M.; Caceres, J.F.; O’connell, M.A. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009, 28, 3145–3156. [Google Scholar] [CrossRef]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef]
- Nemlich, Y.; Greenberg, E.; Ortenberg, R.; Besser, M.J.; Barshack, I.; Jacob-Hirsch, J.; Jacoby, E.; Eyal, E.; Rivkin, L.; Prieto, V.G. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J. Clin. Investig. 2013, 123, 2703–2718. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.; Myung, S.J.; Chang, S. ADAR1 and MicroRNA; A Hidden Crosstalk in Cancer. Int. J. Mol. Sci. 2017, 18, 799. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, G.; Zhang, L.; Zhang, J.; Zhang, J.; Wang, Q.; Billiar, T.R. ADAR1 suppresses the activation of cytosolic RNA-sensing signaling pathways to protect the liver from ischemia/reperfusion injury. Sci. Rep. 2016, 6, 20248. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shoshan, S.O.; Kagan, P.; Sultan, M.; Barabash, Z.; Dor, C.; Jacob-Hirsch, J.; Harmelin, A.; Pappo, O.; Marcu-Malina, V.; Ben-Ari, Z.; et al. ADAR1 deletion induces NFkappaB and interferon signaling dependent liver inflammation and fibrosis. RNA Biol. 2017, 14, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ding, J.; Zhou, X.; Chen, G.; Liu, S.F. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J. Exp. Med. 2008, 205, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Verjans, R.; Peters, T.; Beaumont, F.J.; van Leeuwen, R.; van Herwaarden, T.; Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 family counteracts myocardial fibrosis in pressure overload-induced heart failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef]
- Orecchini, E.; Doria, M.; Michienzi, A.; Giuliani, E.; Vassena, L.; Ciafre, S.A.; Farace, M.G.; Galardi, S. The HIV-1 Tat protein modulates CD4 expression in human T cells through the induction of miR-222. RNA Biol. 2014, 11, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Haurigot, V.; Zhou, S.; Luo, G.; Couto, L.B. Inhibition of hepatitis C virus replication using adeno-associated virus vector delivery of an exogenous anti-hepatitis C virus microRNA cluster. Hepatology 2010, 52, 1877–1887. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Han, L.; Zhang, A.L.; Fu, Y.C.; Yue, X.A.; Wang, G.X.; Jia, Z.F.; Pu, P.Y.; Zhang, Q.Y.; Kang, C.S. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer 2010, 10, 367. [Google Scholar]
- Garofalo, M.; Di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar] [PubMed]
- Griffiths-Jones, S. The microRNA registry. Nucleic Acids Res. 2004, 32, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Sheng, C.; Yang, C.; Chen, L.; Sun, J. Tanshinone IIA inhibits apoptosis in the myocardium by inducing microRNA-152-3p expression and thereby downregulating PTEN. Am. J. Transl. Res. 2016, 8, 3124–3132. [Google Scholar] [PubMed]
- Maisch, B.; Pankuweit, S. Current treatment options in (peri)myocarditis and inflammatory cardiomyopathy. Herz 2012, 37, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Hendry, R.G.; Bilawchuk, L.M.; Marchant, D.J. Targeting matrix metalloproteinase activity and expression for the treatment of viral myocarditis. J. Cardiovasc. Transl. Res. 2014, 7, 212–225. [Google Scholar] [CrossRef]
- Mody, K.P.; Takayama, H.; Landes, E.; Yuzefpolskaya, M.; Colombo, P.C.; Naka, Y.; Jorde, U.P.; Uriel, N. Acute mechanical circulatory support for fulminant myocarditis complicated by cardiogenic shock. J. Cardiovasc. Transl. Res. 2014, 7, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.F.; Ding, Y.J.; Zhang, Z.X.; Wang, Z.F.; Luo, C.L.; Li, B.X.; Shen, Y.W.; Tao, L.Y.; Zhao, Z.Q. MicroRNA-21 regulation of the progression of viral myocarditis to dilated cardiomyopathy. Mol. Med. Rep. 2014, 10, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.F.; Ding, Y.J.; Shen, Y.W.; Xue, A.M.; Xu, H.M.; Luo, C.L.; Li, B.X.; Liu, Y.L.; Zhao, Z.Q. MicroRNA-1 represses Cx43 expression in viral myocarditis. Mol. Cell. Biochem. 2012, 362, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Massilamany, C.; Huber, S.A.; Cunningham, M.W.; Reddy, J. Relevance of molecular mimicry in the mediation of infectious myocarditis. J. Cardiovasc. Transl. Res. 2014, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Sateriale, A.; Budd, R.C.; Huber, S.A.; Buskiewicz, I.A. The role of sex differences in autophagy in the heart during coxsackievirus B3-induced myocarditis. J. Cardiovasc. Transl. Res. 2014, 7, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Antoniak, S.; Mackman, N. Coagulation, protease-activated receptors, and viral myocarditis. J. Cardiovasc. Transl. Res. 2014, 7, 203–211. [Google Scholar] [CrossRef]
- Rabinovici, R.; Kabir, K.; Chen, M.H.; Su, Y.J.; Zhang, D.X.; Luo, X.X.; Yang, J.H. ADAR1 is involved in the development of microvascular lung injury. Circ. Res. 2001, 88, 1066–1071. [Google Scholar] [CrossRef]
- Yang, J.H.; Luo, X.X.; Nie, Y.Z.; Su, Y.J.; Zhao, Q.C.; Kabir, K.; Zhang, D.X.; Rabinovici, R. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 2003, 109, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Gandy, S.Z.; Linnstaedt, S.D.; Muralidhar, S.; Cashman, K.A.; Rosenthal, L.J.; Casey, J.L. RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. J. Virol. 2007, 81, 13544–13551. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.F.; Crews, L.A.; Barrett, C.L.; Chun, H.J.; Court, A.C.; Isquith, J.M.; Zipeto, M.A.; Goff, D.J.; Minden, M.; Sadarangani, A.; et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Li, Y.; Lin, C.H.; Chan, T.H.M.; Chow, R.K.K.; Song, Y.Y.; Liu, M.; Yuan, Y.F.; Fu, L.; Kong, K.L.; et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 2013, 19, 209–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoopengardner, B.; Bhalla, T.; Staber, C.; Reenan, R. Nervous system targets of RNA editing identified by comparative genomics. Science 2003, 301, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef]
- Chunzi, S.; Masayuki, S.; Yusuke, S.; Kazuko, N. Functions of the RNA editing ADAR1 and their relevance to human diseases. Genes 2016, 7, 129. [Google Scholar]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, Y.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2009, 104, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kontos, C.D. Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Bland, J.M.; Kleinhenz, D.J.; Mitchell, P.O.; Walp, E.R.; Sutliff, R.L.; Hart, C.M. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am. J. Respir. Cell Mol. Biol. 2010, 42, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tamura, M.; Yamada, K.M. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J. Cell Biol. 1998, 143, 1375–1383. [Google Scholar] [CrossRef]
- Moon, S.K.; Kim, H.M.; Kim, C.H. PTEN induces G1 cell cycle arrest and inhibits MMP-9 expression via the regulation of NF-kappaB and AP-1 in vascular smooth muscle cells. Arch. Biochem. Biophys. 2004, 421, 267–276. [Google Scholar] [CrossRef]
- Tamguney, T.; Stokoe, D. New insights into PTEN. J. Cell Sci. 2007, 120, 4071–4079. [Google Scholar] [CrossRef] [Green Version]
- Van de Sande, T.; De Schrijver, E.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer Res. 2002, 62, 642–646. [Google Scholar]
- Oudit, G.Y.; Penninger, J.M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 2009, 82, 250–260. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Sun, H.; Kerfant, B.G.; Crackower, M.A.; Penninger, J.M.; Backx, P.H. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 2004, 37, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, W.; Yuan, Y.; Xing, X.; Li, H.; Zhao, G. miR-222 promotes invasion and migration of ovarian carcinoma by targeting PTEN. Oncol. Lett. 2018, 16, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.; Singla, D.K. MicroRNA-1 transfected embryonic stem cells enhance cardiac myocyte differentiation and inhibit apoptosis by modulating the PTEN/Akt pathway in the infarcted heart. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Klingel, K.; Hohenadl, C.; Canu, A.; Albrecht, M.; Seemann, M.; Mall, G.; Kandolf, R. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: Quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. USA 1992, 89, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Kandolf, R.; Selinka, H.; Klingel, K. Pathogenesis of Coxsackievirus B infections. Mol. Biol. Picornaviruses 2002, 67, 405–413. [Google Scholar]
- Rahnefeld, A.; Klingel, K.; Schuermann, A.; Diny, N.L.; Althof, N.; Lindner, A.; Bleienheuft, P.; Savvatis, K.; Respondek, D.; Opitz, E.; et al. Ubiquitin-Like Protein ISG15 (Interferon-Stimulated Gene of 15 kDa) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation 2014, 130, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Yue, Y.; Chen, R.; Xu, W.; Xiong, S. A20 (TNFAIP3) alleviates CVB3-induced myocarditis via inhibiting NF-kappaB signaling. PLoS ONE 2012, 7, e46515. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Gao, X.; Hu, J.; Xie, Y.; Zuo, Y.; Xu, H.; Zhu, S. ADAR1p150 Forms a Complex with Dicer to Promote miRNA-222 Activity and Regulate PTEN Expression in CVB3-Induced Viral Myocarditis. Int. J. Mol. Sci. 2019, 20, 407. https://doi.org/10.3390/ijms20020407
Zhang X, Gao X, Hu J, Xie Y, Zuo Y, Xu H, Zhu S. ADAR1p150 Forms a Complex with Dicer to Promote miRNA-222 Activity and Regulate PTEN Expression in CVB3-Induced Viral Myocarditis. International Journal of Molecular Sciences. 2019; 20(2):407. https://doi.org/10.3390/ijms20020407
Chicago/Turabian StyleZhang, Xincai, Xiangting Gao, Jun Hu, Yuxin Xie, Yuanyi Zuo, Hongfei Xu, and Shaohua Zhu. 2019. "ADAR1p150 Forms a Complex with Dicer to Promote miRNA-222 Activity and Regulate PTEN Expression in CVB3-Induced Viral Myocarditis" International Journal of Molecular Sciences 20, no. 2: 407. https://doi.org/10.3390/ijms20020407
APA StyleZhang, X., Gao, X., Hu, J., Xie, Y., Zuo, Y., Xu, H., & Zhu, S. (2019). ADAR1p150 Forms a Complex with Dicer to Promote miRNA-222 Activity and Regulate PTEN Expression in CVB3-Induced Viral Myocarditis. International Journal of Molecular Sciences, 20(2), 407. https://doi.org/10.3390/ijms20020407