Ceramide and Regulation of Vascular Tone



Abstract
:1. Introduction
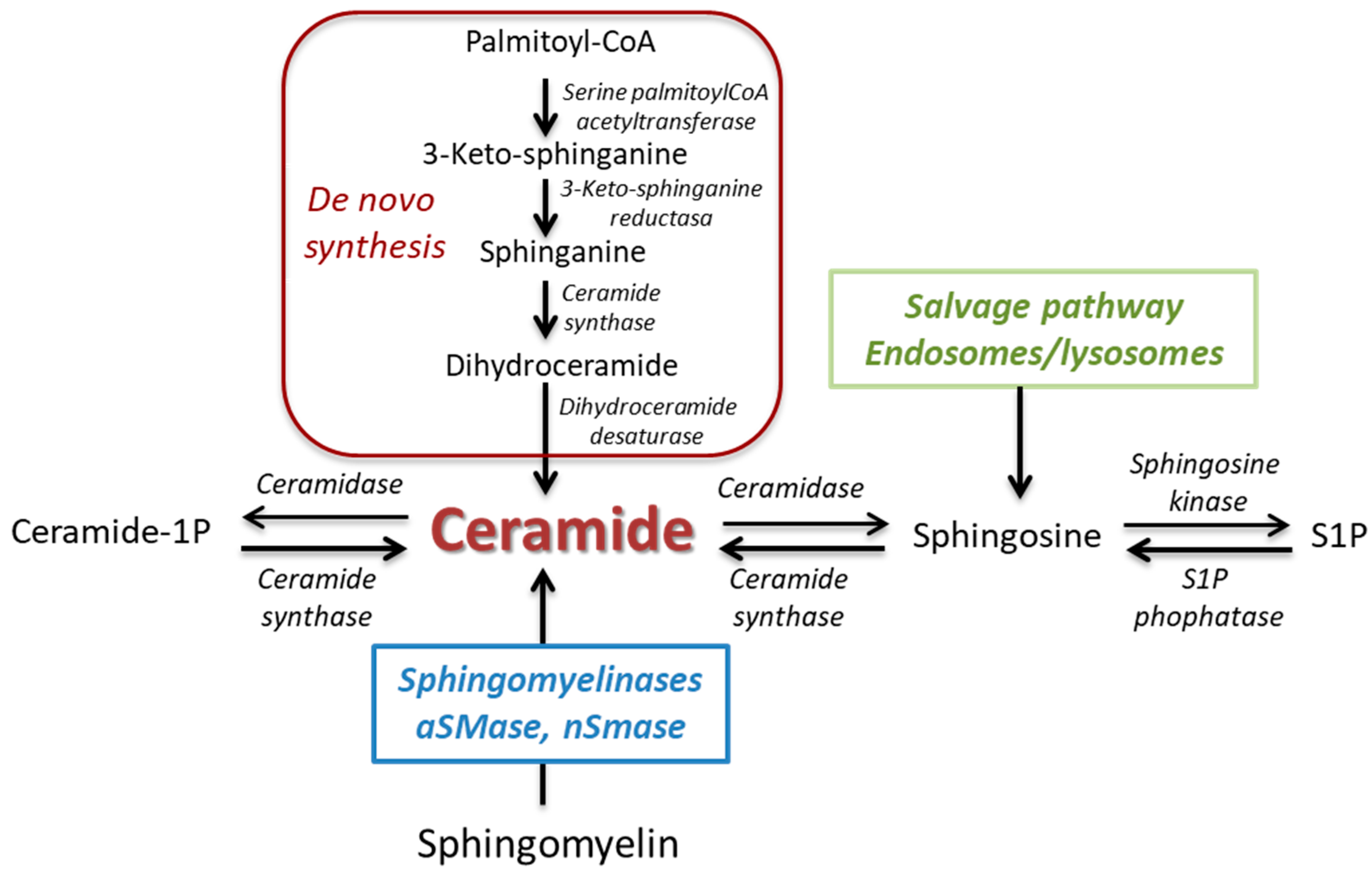
1.1. Synthesis and Metabolism
1.2. The Ceramide/S1P Rheostat
2. Ceramide as Signaling Mediator Regulating Vasomotor Function
2.1. Ceramide-Induced Vasodilation
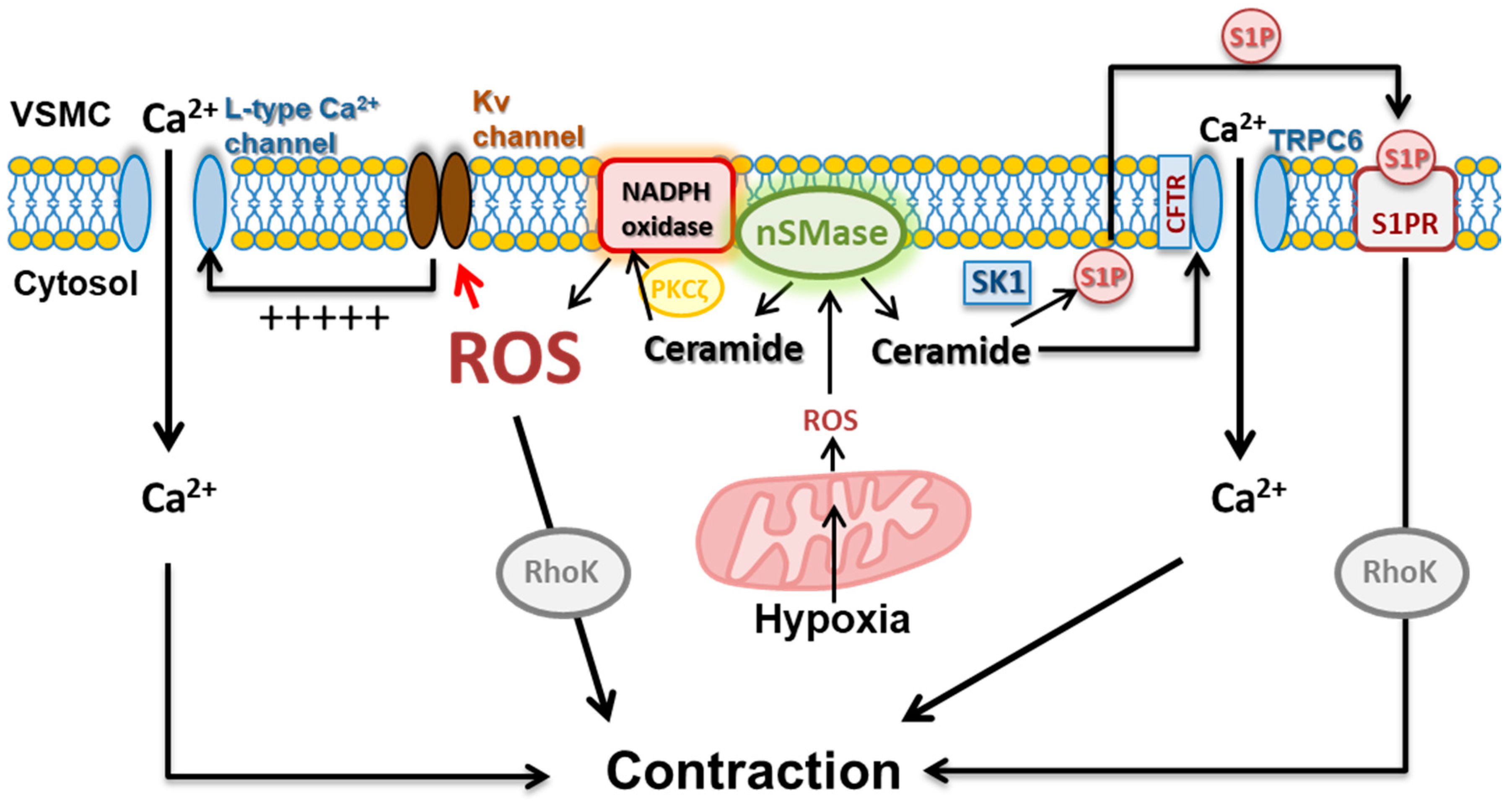
2.2. Ceramide-Induced Vasoconstriction
3. Ceramide and Redox Signaling
3.1. ROS-Induced Ceramide Production
3.2. Ceramide-Induced ROS
3.3. Feedforward Amplifying Mechanism
4. Ceramide and the Endothelium
4.1. Ceramide and Endothelial Dysfunction
4.2. Ceramide and the Endothelial Barrier Function
5. Ceramide as a Key Oxygen- and Mechano-Sensing Mediator
5.1. Ceramide and Oxygen Sensing
5.2. Ceramide and Mechano-Sensing
6. Ceramide and Disease
6.1. Ceramide and Cardiovascular Disease. The Role of Metabolic Syndrome
6.2. Ceramide and Lung Diseases
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R.; Williams, J.B. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: Oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrache, I.; Berdyshev, E.V. Ceramide Signaling and Metabolism in Pathophysiological States of the Lung. Annu. Rev. Physiol. 2016, 78, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, F. Implication of sphingosin-1-phosphate in cardiovascular regulation. Front. Biosci. (Landmark Ed) 2016, 21, 1296–1313. [Google Scholar] [CrossRef] [Green Version]
- Mazurais, D.; Robert, P.; Gout, B.; Berrebi-Bertrand, I.; Laville, M.P.; Calmels, T. Cell type-specific localization of human cardiac S1P receptors. J. Histochem. Cytochem. 2002, 50, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.S.; Bir, A.; Poddar, J.; Sinha, M.; Ganguly, A.; Chakrabarti, S. Ceramide and Sphingosine-1-Phosphate in Cell Death Pathways: Relevance to the Pathogenesis of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1232–1248. [Google Scholar] [CrossRef]
- Kennedy, S.; Kane, K.A.; Pyne, N.J.; Pyne, S. Targeting sphingosine-1-phosphate signalling for cardioprotection. Curr. Opin. Pharmacol. 2009, 9, 194–201. [Google Scholar] [CrossRef]
- Hannun, Y.A. Functions of ceramide in coordinating cellular responses to stress. Science 1996, 274, 1855–1859. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, V.; Diaz-Villanueva, J.F.; Galindo-Hernandez, O.; Martinez-Navarro, I.; Hurtado-Ureta, G.; Perez-Arias, A.A. Ceramide Metabolism Balance, a Multifaceted Factor in Critical Steps of Breast Cancer Development. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Li, P.L.; Gulbins, E. Bioactive Lipids and Redox Signaling: Molecular Mechanism and Disease Pathogenesis. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, J.W.; Donato, L.J.; Bryant, S.C.; Baudhuin, L.M.; Berger, P.B.; Jaffe, A.S. Plasma Ceramides. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1933–1939. [Google Scholar] [CrossRef]
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.T.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat. Med. 2005, 11, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibboel, J.; Reiss, I.; de Jongste, J.C.; Post, M. Sphingolipids in lung growth and repair. Chest 2014, 145, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Trayssac, M.; Hannun, Y.A.; Obeid, L.M. Role of sphingolipids in senescence: Implication in aging and age-related diseases. J. Clin. Investig. 2018, 128, 2702–2712. [Google Scholar] [CrossRef] [PubMed]
- Fenger, M.; Linneberg, A.; Jorgensen, T.; Madsbad, S.; Sobye, K.; Eugen-Olsen, J.; Jeppesen, J. Genetics of the ceramide/sphingosine-1-phosphate rheostat in blood pressure regulation and hypertension. BMC Genet. 2011, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Michel, T. Sphingosine-1-phosphate and modulation of vascular tone. Cardiovasc. Res. 2009, 82, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levkau, B. Sphingosine-1-phosphate in the regulation of vascular tone: A finely tuned integration system of S1P sources, receptors, and vascular responsiveness. Circ. Res. 2008, 103, 231–233. [Google Scholar] [CrossRef]
- Kerage, D.; Brindley, D.N.; Hemmings, D.G. Review: Novel insights into the regulation of vascular tone by sphingosine 1-phosphate. Placenta 2014, 35, S86–S92. [Google Scholar] [CrossRef]
- Czyborra, P.; Saxe, M.; Fetscher, C.; Meyer Zu Heringdorf, D.; Herzig, S.; Jakobs, K.H.; Michel, M.C.; Bischoff, A. Transient relaxation of rat mesenteric microvessels by ceramides. Br. J. Pharmacol. 2002, 135, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Johns, D.G.; Jin, J.S.; Webb, R.C. The role of the endothelium in ceramide-induced vasodilation. Eur. J. Pharmacol. 1998, 349, R9–R10. [Google Scholar] [CrossRef]
- Zheng, T.; Li, W.; Wang, J.; Altura, B.T.; Altura, B.M. C2-ceramide attenuates phenylephrine-induced vasoconstriction and elevation in [Ca2+]i in rat aortic smooth muscle. Lipids 1999, 34, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.J.; Ahn, D.S.; Cho, Y.E.; Morgan, K.G.; Lee, Y.H. C2-ceramide induces vasodilation in phenylephrine-induced pre-contracted rat thoracic aorta: Role of RhoA/Rho-kinase and intracellular Ca2+ concentration. Naunyn Schmiedebergs Arch. Pharmacol. 2005, 372, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Zhang, D.X.; Zou, A.P.; Campbell, W.B. Effect of ceramide on KCa channel activity and vascular tone in coronary arteries. Hypertension 1999, 33, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Li, W.; Wang, J.; Altura, B.T.; Altura, B.M. Sphingomyelinase and ceramide analogs induce contraction and rises in [Ca(2+)](i) in canine cerebral vascular muscle. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1421–H1428. [Google Scholar] [CrossRef] [PubMed]
- Frazziano, G.; Moreno, L.; Moral-Sanz, J.; Menendez, C.; Escolano, L.; Gonzalez, C.; Villamor, E.; Alvarez-Sala, J.L.; Cogolludo, A.L.; Perez-Vizcaino, F. Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J. Cell Physiol. 2011, 226, 2633–2640. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Moreno, L.; Frazziano, G.; Moral-Sanz, J.; Menendez, C.; Castaneda, J.; Gonzalez, C.; Villamor, E.; Perez-Vizcaino, F. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 2009, 82, 296–302. [Google Scholar] [CrossRef]
- Moral-Sanz, J.; Gonzalez, T.; Menendez, C.; David, M.; Moreno, L.; Macias, A.; Cortijo, J.; Valenzuela, C.; Perez-Vizcaino, F.; Cogolludo, A. Ceramide inhibits Kv currents and contributes to TP-receptor-induced vasoconstriction in rat and human pulmonary arteries. Am. J. Physiol. Cell Physiol. 2011, 301, C186–C194. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Moral-Sanz, J.; Morales-Cano, D.; Barreira, B.; Moreno, E.; Ferrarini, A.; Pandolfi, R.; Ruperez, F.J.; Cortijo, J.; Sanchez-Luna, M.; et al. Ceramide mediates acute oxygen sensing in vascular tissues. Antioxid. Redox Signal. 2014, 20, 1–14. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Li, L.; Liu, P.; Wei, Y.; Qian, Z. Ceramide enhances COX-2 expression and VSMC contractile hyperreactivity via ER stress signal activation. Vasc. Pharmacol. 2017, 96–98, 26–32. [Google Scholar] [CrossRef]
- Pandolfi, R.; Barreira, B.; Moreno, E.; Lara-Acedo, V.; Morales-Cano, D.; Martinez-Ramas, A.; de Olaiz Navarro, B.; Herrero, R.; Lorente, J.A.; Cogolludo, A.; et al. Role of acid sphingomyelinase and IL-6 as mediators of endotoxin-induced pulmonary vascular dysfunction. Thorax 2017, 72, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Zou, A.P.; Li, P.L. Ceramide reduces endothelium-dependent vasodilation by increasing superoxide production in small bovine coronary arteries. Circ. Res. 2001, 88, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Faraci, F.M. Ceramide-induced impairment of endothelial function is prevented by CuZn superoxide dismutase overexpression. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.S.; Tsai, C.S.; Si, X.; Webb, R.C. Endothelium dependent and independent relaxations induced by ceramide in vascular smooth muscles. Chin. J. Physiol. 1999, 42, 47–51. [Google Scholar] [PubMed]
- Mogami, K.; Kishi, H.; Kobayashi, S. Sphingomyelinase causes endothelium-dependent vasorelaxation through endothelial nitric oxide production without cytosolic Ca(2+) elevation. FEBS Lett. 2005, 579, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.G.; Osborn, H.; Webb, R.C. Ceramide: A novel cell signaling mechanism for vasodilation. Biochem. Biophys. Res. Commun. 1997, 237, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Li, W.; Altura, B.T.; Altura, B.M. C2-ceramide attenuates prostaglandin F2alpha-induced vasoconstriction and elevation of [Ca2+]i in canine cerebral vascular smooth muscle. Neurosci. Lett. 1998, 256, 113–116. [Google Scholar] [CrossRef]
- Ishii, T.; Warabi, E.; Siow, R.C.M.; Mann, G.E. Sequestosome1/p62: A regulator of redox-sensitive voltage-activated potassium channels, arterial remodeling, inflammation, and neurite outgrowth. Free Radic Biol. Med. 2013, 65, 102–116. [Google Scholar] [CrossRef]
- Moreno, L.; Frazziano, G.; Cogolludo, A.; Cobeno, L.; Tamargo, J.; Perez-Vizcaino, F. Role of protein kinase Czeta and its adaptor protein p62 in voltage-gated potassium channel modulation in pulmonary arteries. Mol. Pharmacol. 2007, 72, 1301–1309. [Google Scholar] [CrossRef]
- Tabeling, C.; Yu, H.; Wang, L.; Ranke, H.; Goldenberg, N.M.; Zabini, D.; Noe, E.; Krauszman, A.; Gutbier, B.; Yin, J.; et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2015, 112, E1614–E1623. [Google Scholar] [CrossRef] [Green Version]
- Bautista-Perez, R.; del Valle-Mondragon, L.; Cano-Martinez, A.; Perez-Mendez, O.; Escalante, B.; Franco, M. Involvement of neutral sphingomyelinase in the angiotensin II signaling pathway. Am. J. Physiol. Renal Physiol. 2015, 308, F1178–F1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldosari, S.; Awad, M.; Harrington, E.O.; Sellke, F.W.; Abid, M.R. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Touyz, R.M. Oxidative stress and hypertension: Current concepts. Curr. Hypertens Rep. 2010, 12, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Dulak-Lis, M.; Tsiropoulou, S.; Harvey, A.; Briones, A.M.; Touyz, R.M. Oxidative stress and human hypertension: Vascular mechanisms, biomarkers, and novel therapies. Can. J. Cardiol. 2015, 31, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vizcaino, F.; Cogolludo, A.; Moreno, L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir. Physiol. Neurobiol. 2010, 174, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Knock, G.A.; Ward, J.P. Redox regulation of protein kinases as a modulator of vascular function. Antioxid. Redox Signal. 2011, 15, 1531–1547. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Frazziano, G.; Cobeno, L.; Moreno, L.; Lodi, F.; Villamor, E.; Tamargo, J.; Perez-Vizcaino, F. Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann. N. Y. Acad. Sci. 2006, 1091, 41–51. [Google Scholar] [CrossRef]
- Bao, J.X.; Su, Y.T.; Cheng, Y.P.; Zhang, H.J.; Xie, X.P.; Chang, Y.M. Vascular sphingolipids in physiological and pathological adaptation. Front. Biosci. 2016, 21, 1168–1186. [Google Scholar]
- Dumitru, C.A.; Zhang, Y.; Li, X.; Gulbins, E. Ceramide: A novel player in reactive oxygen species-induced signaling? Antioxid. Redox Signal. 2007, 9, 1535–1540. [Google Scholar] [CrossRef]
- Li, P.L.; Zhang, Y. Cross talk between ceramide and redox signaling: Implications for endothelial dysfunction and renal disease. Handb. Exp. Pharmacol. 2013, 171–197. [Google Scholar] [CrossRef]
- Li, X.; Becker, K.A.; Zhang, Y. Ceramide in redox signaling and cardiovascular diseases. Cell Physiol. Biochem. 2010, 26, 41–48. [Google Scholar] [CrossRef]
- Martin, D.; Salinas, M.; Fujita, N.; Tsuruo, T.; Cuadrado, A. Ceramide and reactive oxygen species generated by H2O2 induce caspase-3-independent degradation of Akt/protein kinase B. J. Biol. Chem. 2002, 277, 42943–42952. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Edmunds, T.; Baker-Malcolm, J.; Karey, K.P.; Estes, S.; Schwarz, C.; Hughes, H.; Van Patten, S.M. Activation of human acid sphingomyelinase through modification or deletion of C-terminal cysteine. J. Biol. Chem. 2003, 278, 32744–32752. [Google Scholar] [CrossRef] [PubMed]
- Scheel-Toellner, D.; Wang, K.; Craddock, R.; Webb, P.R.; McGettrick, H.M.; Assi, L.K.; Parkes, N.; Clough, L.E.; Gulbins, E.; Salmon, M.; et al. Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood 2004, 104, 2557–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsyupko, A.N.; Dudnik, L.B.; Evstigneeva, R.P.; Alessenko, A.V. Effects of reduced and oxidized glutathione on sphingomyelinase activity and contents of sphingomyelin and lipid peroxidation products in murine liver. Biochemistry 2001, 66, 1028–1034. [Google Scholar] [PubMed]
- Bezombes, C.; Plo, I.; Mansat-De Mas, V.; Quillet-Mary, A.; Negre-Salvayre, A.; Laurent, G.; Jaffrezou, J.P. Oxidative stress-induced activation of Lyn recruits sphingomyelinase and is requisite for its stimulation by Ara-C. FASEB J. 2001, 15, 1583–1585. [Google Scholar] [CrossRef] [PubMed]
- Jesko, H.; Stepien, A.; Lukiw, W.J.; Strosznajder, R.P. The Cross-Talk Between Sphingolipids and Insulin-Like Growth Factor Signaling: Significance for Aging and Neurodegeneration. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Schuchman, E.H. Ceramide and Ischemia/Reperfusion Injury. J. Lipids 2018, 2018, 3646725. [Google Scholar] [CrossRef]
- Li, C.; Wu, Y.; Riehle, A.; Orian-Rousseau, V.; Zhang, Y.; Gulbins, E.; Grassme, H. Regulation of Staphylococcus aureus Infection of Macrophages by CD44, Reactive Oxygen Species, and Acid Sphingomyelinase. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef]
- Charruyer, A.; Grazide, S.; Bezombes, C.; Muller, S.; Laurent, G.; Jaffrezou, J.P. UV-C light induces raft-associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J. Biol. Chem. 2005, 280, 19196–19204. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Gulbins, E. TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene 2006, 25, 5612–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinq-Frais, C.; Coatrieux, C.; Grazide, M.H.; Hannun, Y.A.; Negre-Salvayre, A.; Salvayre, R.; Auge, N. A signaling cascade mediated by ceramide, src and PDGFRbeta coordinates the activation of the redox-sensitive neutral sphingomyelinase-2 and sphingosine kinase-1. Biochim. Biophys. Acta 2013, 1831, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Castillo, S.S.; Goldkorn, T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem. Biophys. Res. Commun. 2006, 344, 900–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camare, C.; Auge, N.; Pucelle, M.; Saint-Lebes, B.; Grazide, M.H.; Negre-Salvayre, A.; Salvayre, R. The neutral sphingomyelinase-2 is involved in angiogenic signaling triggered by oxidized LDL. Free Radic. Biol. Med. 2016, 93, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Junk, P.; Huwiler, A.; Burkhardt, C.; Wallerath, T.; Pfeilschifter, J.; Forstermann, U. Dual effect of ceramide on human endothelial cells: Induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation 2002, 106, 2250–2256. [Google Scholar] [CrossRef] [PubMed]
- Lecour, S.; Van der Merwe, E.; Opie, L.H.; Sack, M.N. Ceramide attenuates hypoxic cell death via reactive oxygen species signaling. J. Cardiovasc. Pharmacol. 2006, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Colell, A.; Mari, M.; Morales, A.; Fernandez-Checa, J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef]
- Corda, S.; Laplace, C.; Vicaut, E.; Duranteau, J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am. J. Respir. Cell Mol. Biol. 2001, 24, 762–768. [Google Scholar] [CrossRef]
- Safiedeen, Z.; Rodriguez-Gomez, I.; Vergori, L.; Soleti, R.; Vaithilingam, D.; Douma, I.; Agouni, A.; Leiber, D.; Dubois, S.; Simard, G.; et al. Temporal Cross Talk Between Endoplasmic Reticulum and Mitochondria Regulates Oxidative Stress and Mediates Microparticle-Induced Endothelial Dysfunction. Antioxid. Redox Signal. 2017, 26, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Freed, J.K.; Beyer, A.M.; LoGiudice, J.A.; Hockenberry, J.C.; Gutterman, D.D. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ. Res. 2014, 115, 525–532. [Google Scholar] [CrossRef]
- Bhunia, A.K.; Han, H.; Snowden, A.; Chatterjee, S. Redox-regulated signaling by lactosylceramide in the proliferation of human aortic smooth muscle cells. J. Biol. Chem. 1997, 272, 15642–15649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Zou, A.P.; Li, P.L. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H605–H612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Yi, F.; Jin, S.; Xia, M.; Chen, Q.Z.; Gulbins, E.; Li, P.L. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid. Redox Signal. 2007, 9, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, P.L. Membrane raft redox signalosomes in endothelial cells. Free Radic. Res. 2010, 44, 831–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffrezou, J.P.; Maestre, N.; de Mas-Mansat, V.; Bezombes, C.; Levade, T.; Laurent, G. Positive feedback control of neutral sphingomyelinase activity by ceramide. FASEB J. 1998, 12, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Villamor, E. Mechanisms controlling vascular tone in pulmonary arterial hypertension: Implications for vasodilator therapy. Pharmacology 2007, 79, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Barsacchi, R.; Perrotta, C.; Bulotta, S.; Moncada, S.; Borgese, N.; Clementi, E. Activation of endothelial nitric-oxide synthase by tumor necrosis factor-alpha: A novel pathway involving sequential activation of neutral sphingomyelinase, phosphatidylinositol-3’ kinase, and Akt. Mol. Pharmacol. 2003, 63, 886–895. [Google Scholar] [CrossRef]
- De Palma, C.; Meacci, E.; Perrotta, C.; Bruni, P.; Clementi, E. Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: A novel pathway relevant to the pathophysiology of endothelium. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Symons, J.D.; Abel, E.D. Lipotoxicity contributes to endothelial dysfunction: A focus on the contribution from ceramide. Rev. Endocr. Metab. Disord. 2013, 14, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.R.; Visioli, F.; Frei, B.; Hagen, T.M. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: Evidence for a novel mechanism involving sphingomyelinase and ceramide-activated phosphatase 2A. Aging Cell 2006, 5, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yin, J.; Baumgartner, W.; Samapati, R.; Solymosi, E.A.; Reppien, E.; Kuebler, W.M.; Uhlig, S. Platelet-activating factor reduces endothelial nitric oxide production: Role of acid sphingomyelinase. Eur.Respir. J. 2010, 36, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, N.A.; Brovkovych, V.; Allen, S.E.; John, T.A.; Shajahan, A.N.; Tiruppathi, C.; Vogel, S.M.; Skidgel, R.A.; Malik, A.B.; Minshall, R.D. Novel mechanism of endothelial nitric oxide synthase activation mediated by caveolae internalization in endothelial cells. Circ. Res. 2006, 99, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Song, P.; Xu, J.; Zhang, M.; Zou, M.H. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 9777–9788. [Google Scholar] [CrossRef]
- Bharath, L.P.; Ruan, T.; Li, Y.; Ravindran, A.; Wan, X.; Nhan, J.K.; Walker, M.L.; Deeter, L.; Goodrich, R.; Johnson, E.; et al. Ceramide-Initiated Protein Phosphatase 2A Activation Contributes to Arterial Dysfunction In Vivo. Diabetes 2015, 64, 3914–3926. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Holland, W.L.; Wilson, L.; Tanner, J.M.; Kearns, D.; Cahoon, J.M.; Pettey, D.; Losee, J.; Duncan, B.; Gale, D.; et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 2012, 61, 1848–1859. [Google Scholar] [CrossRef]
- Jin, S.; Zhang, Y.; Yi, F.; Li, P.L. Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 485–490. [Google Scholar] [CrossRef]
- Schutze, S.; Berkovic, D.; Tomsing, O.; Unger, C.; Kronke, M. Tumor necrosis factor induces rapid production of 1’2’diacylglycerol by a phosphatidylcholine-specific phospholipase C. J. Exp. Med. 1991, 174, 975–988. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorbara, M.T.; Girardin, S.E. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011, 21, 558–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.B.; Hunt, W.R.; Noe, J.E.; Rush, N.I.; Schweitzer, K.S.; Leece, T.C.; Moldobaeva, A.; Wagner, E.M.; Dudek, S.M.; Poirier, C.; et al. Loss of cystic fibrosis transmembrane conductance regulator impairs lung endothelial cell barrier function and increases susceptibility to microvascular damage from cigarette smoke. Pulm. Circ. 2014, 4, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.S.; Hatoum, H.; Brown, M.B.; Gupta, M.; Justice, M.J.; Beteck, B.; Van Demark, M.; Gu, Y.; Presson, R.G., Jr.; Hubbard, W.C.; et al. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: Role of oxidative stress and ceramides. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L836–L846. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.; Erfinanda, L.; Bartz, C.; Kuebler, W.M. Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 2018. [Google Scholar] [CrossRef]
- Goggel, R.; Winoto-Morbach, S.; Vielhaber, G.; Imai, Y.; Lindner, K.; Brade, L.; Brade, H.; Ehlers, S.; Slutsky, A.S.; Schutze, S.; et al. PAF-mediated pulmonary edema: A new role for acid sphingomyelinase and ceramide. Nat. Med. 2004, 10, 155–160. [Google Scholar] [CrossRef]
- Kuebler, W.M.; Yang, Y.; Samapati, R.; Uhlig, S. Vascular barrier regulation by PAF, ceramide, caveolae, and NO - an intricate signaling network with discrepant effects in the pulmonary and systemic vasculature. Cell Physiol. Biochem. 2010, 26, 29–40. [Google Scholar] [CrossRef]
- Sommer, N.; Huttemann, M.; Pak, O.; Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; et al. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circ. Res. 2017, 121, 424–438. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Schumacker, P.T. Hypoxic pulmonary vasoconstriction: Redox events in oxygen sensing. J. Appl. Physiol. 2005, 98, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.K.; Lopez-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen-sensing mechanisms. N. Engl. J. Med. 2005, 353, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, P.I.; Robertson, T.P.; Knock, G.A.; Becker, S.; Lewis, T.H.; Snetkov, V.; Ward, J.P. Hypoxic pulmonary vasoconstriction: Mechanisms and controversies. J. Physiol. 2006, 570, 53–58. [Google Scholar] [CrossRef]
- Weir, E.K.; Archer, S.L. The mechanism of acute hypoxic pulmonary vasoconstriction: The tale of two channels. FASEB J. 1995, 9, 183–189. [Google Scholar] [CrossRef]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 2001, 88, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Dietrich, A.; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Mederos y Schnitzler, M.; Ghofrani, H.A.; et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. USA 2006, 103, 19093–19098. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Marks, J.D.; Guzy, R.; Mungai, P.T.; Schriewer, J.; Dokic, D.; Schumacker, P.T. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 2010, 106, 526–535. [Google Scholar] [CrossRef]
- Bourbon, N.A.; Yun, J.; Kester, M. Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J. Biol. Chem. 2000, 275, 35617–35623. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Bosca, L.; Tamargo, J.; Perez-Vizcaino, F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: Role of protein kinase Czeta. Circ. Res. 2003, 93, 656–663. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Lodi, F.; Tamargo, J.; Perez-Vizcaino, F. Postnatal maturational shift from PKCzeta and voltage-gated K+ channels to RhoA/Rho kinase in pulmonary vasoconstriction. Cardiovasc. Res. 2005, 66, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Robertson, T.P.; Dipp, M.; Ward, J.P.; Aaronson, P.I.; Evans, A.M. Inhibition of sustained hypoxic vasoconstriction by Y-27632 in isolated intrapulmonary arteries and perfused lung of the rat. Br. J. Pharmacol. 2000, 131, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.L.; Gordienko, D.V.; Yuill, K.H.; Smirnov, S.V. Cellular localization of mitochondria contributes to Kv channel-mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L347–L360. [Google Scholar] [CrossRef] [Green Version]
- Clyman, R.I. Mechanisms regulating the ductus arteriosus. Biol. Neonate 2006, 89, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.; Rebeyka, I.; Bateson, J.; Olley, P.; Puttagunta, L.; Archer, S. Voltage-gated potassium channels in human ductus arteriosus. Lancet 2000, 356, 134–137. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Rebeyka, I.; Wu, X.; Nsair, A.; Thebaud, B.; Hashimoto, K.; Dyck, J.R.; Haromy, A.; Harry, G.; Barr, A.; et al. O2 sensing in the human ductus arteriosus: Regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ. Res. 2002, 91, 478–486. [Google Scholar] [CrossRef]
- Cogolludo, A.L.; Moral-Sanz, J.; van der Sterren, S.; Frazziano, G.; van Cleef, A.N.; Menendez, C.; Zoer, B.; Moreno, E.; Roman, A.; Perez-Vizcaino, F.; et al. Maturation of O2 sensing and signaling in the chicken ductus arteriosus. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L619–L630. [Google Scholar] [CrossRef]
- Olschewski, A.; Hong, Z.; Peterson, D.A.; Nelson, D.P.; Porter, V.A.; Weir, E.K. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L15–L22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Z.; Hong, F.; Olschewski, A.; Cabrera, J.A.; Varghese, A.; Nelson, D.P.; Weir, E.K. Role of store-operated calcium channels and calcium sensitization in normoxic contraction of the ductus arteriosus. Circulation 2006, 114, 1372–1379. [Google Scholar] [CrossRef]
- Kajimoto, H.; Hashimoto, K.; Bonnet, S.N.; Haromy, A.; Harry, G.; Moudgil, R.; Nakanishi, T.; Rebeyka, I.; Thebaud, B.; Michelakis, E.D.; et al. Oxygen activates the Rho/Rho-kinase pathway and induces RhoB and ROCK-1 expression in human and rabbit ductus arteriosus by increasing mitochondria-derived reactive oxygen species: A newly recognized mechanism for sustaining ductal constriction. Circulation 2007, 115, 1777–1788. [Google Scholar] [CrossRef]
- Czarny, M.; Liu, J.; Oh, P.; Schnitzer, J.E. Transient mechanoactivation of neutral sphingomyelinase in caveolae to generate ceramide. J. Biol. Chem. 2003, 278, 4424–4430. [Google Scholar] [CrossRef] [PubMed]
- Czarny, M.; Schnitzer, J.E. Neutral sphingomyelinase inhibitor scyphostatin prevents and ceramide mimics mechanotransduction in vascular endothelium. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1344–H1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, J.K.; Durand, M.J.; Hoffmann, B.R.; Densmore, J.C.; Greene, A.S.; Gutterman, D.D. Mitochondria-regulated formation of endothelium-derived extracellular vesicles shifts the mediator of flow-induced vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1096–H1104. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H. Acid ceramidase and the treatment of ceramide diseases: The expanding role of enzyme replacement therapy. Biochim. Biophys. Acta 2016, 1862, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Gilbert-Barness, E. Cardiovascular involvement in metabolic diseases. Pediatr. Pathol. Mol. Med. 2002, 21, 93–136. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.C.; Wallington-Beddoe, C.T.; Powell, J.A.; Pitson, S.M. Targeting sphingolipid metabolism as an approach for combination therapies in haematological malignancies. Cell Death Discov. 2018, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Dinoff, A.; Herrmann, N.; Lanctot, K.L. Ceramides and depression: A systematic review. J. Affect. Disord. 2017, 213, 35–43. [Google Scholar] [CrossRef]
- Ong, W.Y.; Herr, D.R.; Farooqui, T.; Ling, E.A.; Farooqui, A.A. Role of sphingomyelinases in neurological disorders. Expert Opin. Ther. Targets 2015, 19, 1725–1742. [Google Scholar] [CrossRef]
- Baker, J.E.; Boudreau, R.M.; Seitz, A.P.; Caldwell, C.C.; Gulbins, E.; Edwards, M.J. Sphingolipids and Innate Immunity: A New Approach to Infection in the Post-Antibiotic Era? Surg. Infect. 2018. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.C. The Role of Sphingolipids on Innate Immunity to Intestinal Salmonella Infection. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Norris, G.H.; Blesso, C.N. Dietary and Endogenous Sphingolipid Metabolism in Chronic Inflammation. Nutrients 2017, 9. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J. Intern. Med. 2018, 284, 478–491. [Google Scholar] [CrossRef] [Green Version]
- Cruciani-Guglielmacci, C.; Lopez, M.; Campana, M.; le Stunff, H. Brain Ceramide Metabolism in the Control of Energy Balance. Front. Physiol. 2017, 8, 787. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef]
- Meikle, P.J.; Summers, S.A. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat. Rev. Endocrinol. 2017, 13, 79–91. [Google Scholar] [CrossRef]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [Green Version]
- Summers, S.A. Could Ceramides Become the New Cholesterol? Cell Metab. 2018, 27, 276–280. [Google Scholar] [CrossRef]
- Tippetts, T.S.; Holland, W.L.; Summers, S.A. The ceramide ratio: A predictor of cardiometabolic risk. J. Lipid Res. 2018, 59, 1549–1550. [Google Scholar] [CrossRef] [PubMed]
- Borodzicz, S.; Czarzasta, K.; Kuch, M.; Cudnoch-Jedrzejewska, A. Sphingolipids in cardiovascular diseases and metabolic disorders. Lipids Health Dis. 2015, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Robciuc, A.; Hyotylainen, T.; Jauhiainen, M.; Holopainen, J.M. Ceramides in the pathophysiology of the anterior segment of the eye. Curr. Eye Res. 2013, 38, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ganguli-Indra, G.; Indra, A.K. Lipidomic analysis of epidermal lipids: A tool to predict progression of inflammatory skin disease in humans. Expert Rev. Proteomics 2016, 13, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Borodzicz, S.; Rudnicka, L.; Mirowska-Guzel, D.; Cudnoch-Jedrzejewska, A. The role of epidermal sphingolipids in dermatologic diseases. Lipids Health Dis. 2016, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, K.; Ekroos, K.; Suoniemi, M.; Kauhanen, D.; Sylvanne, T.; Hurme, R.; Gouni-Berthold, I.; Berthold, H.K.; Kleber, M.E.; Laaksonen, R.; et al. Molecular lipids identify cardiovascular risk and are efficiently lowered by simvastatin and PCSK9 deficiency. J. Clin. Endocrinol. Metab. 2014, 99, E45–E52. [Google Scholar] [CrossRef] [PubMed]
- Havulinna, A.S.; Sysi-Aho, M.; Hilvo, M.; Kauhanen, D.; Hurme, R.; Ekroos, K.; Salomaa, V.; Laaksonen, R. Circulating Ceramides Predict Cardiovascular Outcomes in the Population-Based FINRISK 2002 Cohort. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2424–2430. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.D.; Toledo, E.; Hruby, A.; Rosner, B.A.; Willett, W.C.; Sun, Q.; Razquin, C.; Zheng, Y.; Ruiz-Canela, M.; Guasch-Ferre, M.; et al. Plasma Ceramides, Mediterranean Diet, and Incident Cardiovascular Disease in the PREDIMED Trial (Prevencion con Dieta Mediterranea). Circulation 2017, 135, 2028–2040. [Google Scholar] [CrossRef]
- Mantovani, A.; Bonapace, S.; Lunardi, G.; Salgarello, M.; Dugo, C.; Canali, G.; Byrne, C.D.; Gori, S.; Barbieri, E.; Targher, G. Association between plasma ceramides and inducible myocardial ischemia in patients with established or suspected coronary artery disease undergoing myocardial perfusion scintigraphy. Metabolism 2018, 85, 305–312. [Google Scholar] [CrossRef]
- Anroedh, S.; Hilvo, M.; Akkerhuis, K.M.; Kauhanen, D.; Koistinen, K.; Oemrawsingh, R.; Serruys, P.; van Geuns, R.J.; Boersma, E.; Laaksonen, R.; et al. Plasma concentrations of molecular lipid species predict long-term clinical outcome in coronary artery disease patients. J. Lipid Res. 2018, 59, 1729–1737. [Google Scholar] [CrossRef]
- Laaksonen, R.; Ekroos, K.; Sysi-Aho, M.; Hilvo, M.; Vihervaara, T.; Kauhanen, D.; Suoniemi, M.; Hurme, R.; Marz, W.; Scharnagl, H.; et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J. 2016, 37, 1967–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshehry, Z.H.; Mundra, P.A.; Barlow, C.K.; Mellett, N.A.; Wong, G.; McConville, M.J.; Simes, J.; Tonkin, A.M.; Sullivan, D.R.; Barnes, E.H.; et al. Plasma Lipidomic Profiles Improve on Traditional Risk Factors for the Prediction of Cardiovascular Events in Type 2 Diabetes Mellitus. Circulation 2016, 134, 1637–1650. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, L.P.; Tan, S.H.; Ow, G.S.; Tang, Z.; Ching, J.; Kovalik, J.P.; Poh, S.C.; Chin, C.T.; Richards, A.M.; Martinez, E.C.; et al. Plasma Ceramides as Prognostic Biomarkers and Their Arterial and Myocardial Tissue Correlates in Acute Myocardial Infarction. JACC Basic Transl. Sci. 2018, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J. A comprehensive review on metabolic syndrome. Cardiol. Res. Pract. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Summers, S.A. Ceramides—Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Rämö, J.T.; Ripatti, P.; Koskela, J.T.; Kurki, M.; Karjalainen, J.; Hassan, S.; Nunez-Fontarnau, J.; Kiiskinen, T.T.; Söderlund, S. Genetics of human plasma lipidome: Understanding lipid metabolism and its link to diseases beyond traditional lipids. bioRxiv 2018, 457960. [Google Scholar] [CrossRef]
- Wheeler, E.; Leong, A.; Liu, C.T.; Hivert, M.F.; Strawbridge, R.J.; Podmore, C.; Li, M.; Yao, J.; Sim, X.; Hong, J.; et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis. PLoS Med. 2017, 14, e1002383. [Google Scholar] [CrossRef]
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A. Plasma dihydroceramides are diabetes susceptibility biomarker candidates in mice and humans. Cell Rep. 2017, 18, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, R.N.; Yu, C.; Hoofnagle, A.; Hari, N.; Jensen, P.; Fretts, A.M.; Umans, J.G.; Howard, B.V.; Sitlani, C.M.; Siscovick, D.S. Circulating Sphingolipids, Insulin, HOMA-IR and HOMA-B: The Strong Heart Family Study. Diabetes 2018, 68. [Google Scholar] [CrossRef]
- Park, T.S.; Goldberg, I.J. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail Clin. 2012, 8, 633–641. [Google Scholar] [CrossRef]
- Liu, Y.; Neumann, D.; Glatz, J.F.; Luiken, J.J. Molecular mechanism of lipid-induced cardiac insulin resistance and contractile dysfunction. Prostaglandins Leukot. Essent. Fatty Acids 2016, 136, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Summers, S.A. Strong Heart, Low Ceramides. Diabetes 2018, 67, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, S.; Gulbins, E. Sphingolipids in the lungs. Am. J. Respir. Crit. Care Med. 2008, 178, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, S.; Yang, Y. Sphingolipids in acute lung injury. Handb. Exp. Pharmacol. 2013, 227–246. [Google Scholar] [CrossRef]
- Bull, T.M.; Clark, B.; McFann, K.; Moss, M. Pulmonary vascular dysfunction is associated with poor outcomes in patients with acute lung injury. Am. J. Respir. Crit. Care Med. 2010, 182, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Price, L.C.; Wort, S.J.; Finney, S.J.; Marino, P.S.; Brett, S.J. Pulmonary vascular and right ventricular dysfunction in adult critical care: Current and emerging options for management: A systematic literature review. Crit. Care 2010, 14, R169. [Google Scholar] [CrossRef] [PubMed]
- Claus, R.A.; Bunck, A.C.; Bockmeyer, C.L.; Brunkhorst, F.M.; Losche, W.; Kinscherf, R.; Deigner, H.P. Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB J. 2005, 19, 1719–1721. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Tan, G.; Liu, P.; Li, H.; Tang, L.; Huang, L.; Ren, Q. Three plasma metabolite signatures for diagnosing high altitude pulmonary edema. Sci. Rep. 2015, 5, 15126. [Google Scholar] [CrossRef] [Green Version]
- von Bismarck, P.; Wistadt, C.F.; Klemm, K.; Winoto-Morbach, S.; Uhlig, U.; Schutze, S.; Adam, D.; Lachmann, B.; Uhlig, S.; Krause, M.F. Improved pulmonary function by acid sphingomyelinase inhibition in a newborn piglet lavage model. Am. J. Respir. Crit. Care Med. 2008, 177, 1233–1241. [Google Scholar] [CrossRef]
- Yang, J.; Qu, J.M.; Summah, H.; Zhang, J.; Zhu, Y.G.; Jiang, H.N. Protective effects of imipramine in murine endotoxin-induced acute lung injury. Eur. J. Pharmacol. 2010, 638, 128–133. [Google Scholar] [CrossRef]
- Laube, M.; Amann, E.; Uhlig, U.; Yang, Y.; Fuchs, H.W.; Zemlin, M.; Mercier, J.C.; Maier, R.F.; Hummler, H.D.; Uhlig, S.; et al. Inflammatory Mediators in Tracheal Aspirates of Preterm Infants Participating in a Randomized Trial of Inhaled Nitric Oxide. PLoS ONE 2017, 12, e0169352. [Google Scholar] [CrossRef] [PubMed]
- van Mastrigt, E.; Zweekhorst, S.; Bol, B.; Tibboel, J.; van Rosmalen, J.; Samsom, J.N.; Kroon, A.A.; de Jongste, J.C.; Reiss, I.K.M.; Post, M.; et al. Ceramides in tracheal aspirates of preterm infants: Marker for bronchopulmonary dysplasia. PLoS ONE 2018, 13, e0185969. [Google Scholar] [CrossRef] [PubMed]
- Snoek, K.G.; Reiss, I.K.; Tibboel, J.; van Rosmalen, J.; Capolupo, I.; van Heijst, A.; Schaible, T.; Post, M.; Tibboel, D. Sphingolipids in Congenital Diaphragmatic Hernia; Results from an International Multicenter Study. PLoS ONE 2016, 11, e0155136. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilbault, C.; Wojewodka, G.; Saeed, Z.; Hajduch, M.; Matouk, E.; De Sanctis, J.B.; Radzioch, D. Cystic fibrosis fatty acid imbalance is linked to ceramide deficiency and corrected by fenretinide. Am. J. Respir. Cell Mol. Biol. 2009, 41, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Garic, D.; De Sanctis, J.B.; Wojewodka, G.; Houle, D.; Cupri, S.; Abu-Arish, A.; Hanrahan, J.W.; Hajduch, M.; Matouk, E.; Radzioch, D. Fenretinide differentially modulates the levels of long- and very long-chain ceramides by downregulating Cers5 enzyme: Evidence from bench to bedside. J. Mol. Med. 2017, 95, 1053–1064. [Google Scholar] [CrossRef]
- Brodlie, M.; McKean, M.C.; Johnson, G.E.; Gray, J.; Fisher, A.J.; Corris, P.A.; Lordan, J.L.; Ward, C. Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2010, 182, 369–375. [Google Scholar] [CrossRef]
- Lea, S.R.; Metcalfe, H.J.; Plumb, J.; Beerli, C.; Poll, C.; Singh, D.; Abbott-Banner, K.H. Neutral sphingomyelinase-2, acid sphingomyelinase, and ceramide levels in COPD patients compared to controls. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 2139–2147. [Google Scholar] [CrossRef]
- Scarpa, M.C.; Baraldo, S.; Marian, E.; Turato, G.; Calabrese, F.; Saetta, M.; Maestrelli, P. Ceramide expression and cell homeostasis in chronic obstructive pulmonary disease. Respiration 2013, 85, 342–349. [Google Scholar] [CrossRef]
- Bowler, R.P.; Jacobson, S.; Cruickshank, C.; Hughes, G.J.; Siska, C.; Ory, D.S.; Petrache, I.; Schaffer, J.E.; Reisdorph, N.; Kechris, K. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am. J. Respir. Crit. Care Med. 2015, 191, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Younoszai, A.; Miller, J.I.; Kinsella, J.P.; Baker, C.D.; Poindexter, B.B.; Ingram, D.A.; Abman, S.H. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2015, 191, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Galie, N.; Rubin, L.J.; Langleben, D.; Seeger, W.; Domenighetti, G.; Gibbs, S.; Lebrec, D.; Speich, R.; Beghetti, M.; et al. Clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43, 5S–12S. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.S.; Pyne, S.; Macritchie, N.; Pyne, N.J.; Bittman, R. Novel sphingosine-containing analogues selectively inhibit sphingosine kinase (SK) isozymes, induce SK1 proteasomal degradation and reduce DNA synthesis in human pulmonary arterial smooth muscle cells. Medchemcomm 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Gairhe, S.; Joshi, S.R.; Bastola, M.M.; McLendon, J.M.; Oka, M.; Fagan, K.A.; McMurtry, I.F. Sphingosine-1-phosphate is involved in the occlusive arteriopathy of pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Chu, L.; Lin, K.; Granton, E.; Yin, L.; Peng, J.; Hsin, M.; Wu, L.; Yu, A.; Waddell, T.; et al. A Biochemical Approach to Understand the Pathogenesis of Advanced Pulmonary Arterial Hypertension: Metabolomic Profiles of Arginine, Sphingosine-1-Phosphate, and Heme of Human Lung. PLoS ONE 2015, 10, e0134958. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D., Jr.; Tobias, J.D.; Mansour, H.M.; Kirkby, S.; McCoy, K.S.; Daniels, C.J.; Whitson, B.A. Pulmonary hypertension in cystic fibrosis with advanced lung disease. Am. J. Respir. Crit. Care Med. 2014, 190, 898–905. [Google Scholar] [CrossRef]
- Minai, O.A.; Chaouat, A.; Adnot, S. Pulmonary hypertension in COPD: Epidemiology, significance, and management: Pulmonary vascular disease: The global perspective. Chest 2010, 137, 39S–51S. [Google Scholar] [CrossRef]
- Filosto, S.; Castillo, S.; Danielson, A.; Franzi, L.; Khan, E.; Kenyon, N.; Last, J.; Pinkerton, K.; Tuder, R.; Goldkorn, T. Neutral sphingomyelinase 2: A novel target in cigarette smoke-induced apoptosis and lung injury. Am. J. Respir Cell Mol. Biol. 2011, 44, 350–360. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Stimulus | Vascular Effect | Preparation | Mechanism | Ref. |
|---|---|---|---|---|
| C2-ceramide | Transient vasodilation | Rat mesenteric arteries | Activation of Guanylyl cyclase, KCa channels | [20] |
| C2-ceramide | Vasodilation | Rat aorta | Endothelium-derived NO | [21] |
| C2-ceramide | Vasodilation | Rat aorta | Endothelium-independent | [22] |
| C2-ceramide, SMase | Vasodilation | Rat aorta | Inhibition of RhoA/Rho kinase and Ca entry | [23] |
| C2-ceramide | Vasoconstriction | Small bovine coronary arteries | Inhibition of Kca | [24] |
| SMase, C8 and C16-ceramide | Vasoconstriction | canine cerebral arteries | PKC activation and Ca entry | [25] |
| SMase C6-ceramide | Vasoconstriction | Rat pulmonary arteries | PKCζ activation, Rho kinase activation and decreased Ca entry | [26,27] |
| SMase C6-ceramide | Vasoconstriction | Rat and human pulmonary arteries | PKCζ activation and Kv channel inhibition | [28] |
| SMase C6-ceramide | Vasoconstriction | Chicken and human pulmonary arteries and ductus arteriosus | PKCζ activation and Kv channel inhibition | [29] |
| C2-ceramide | Enhanced vasoconstriction | Rat aorta | Endoplasmic reticulum stress | [30] |
| SMase | Reduced vasodilation Increased contraction to 5-HT | Rat pulmonary arteries | IL-6 | [31] |
| C2-ceramide | Attenuated vasodilation | Bovine coronary arteries | Increased ROS | [32] |
| C2-ceramide | Attenuated vasodilation | Mice carotid arteries | Increased ROS | [33] |
| C6, C24:1 and C24:0 ceramide | None | Canine cerebral arteries | [25] |
| Study | Population | Main Findings |
|---|---|---|
| Tarasov et al. 2014 [146] LURIC | Males (258) with coronary artery disease who died within 3 years of follow-up and 187 matched control patients with coronary artery disease who did not die during the follow-up. | Higher plasmatic levels of C16:0, C18:0, C20:0, and C24:1, and lower levels of C24:0 in patients who died. Ratios of C16:0 /C24:0 and C22:0/C24:0 were significantly related to increased risk of death in all subjects and subgroups. C24:0/C24:1 was indicative of a reduced risk of death, regardless of diabetes status. Simvastatin lowered plasma ceramides. |
| Alshehry et al. 2016 [152] ADVANCE | Cohort of 3799 individuals with type 2 diabetes that included 698 patients with cardiovascular events and 355 with cardiovascular death. | Significant association between C24:1 and death. |
| Havulinna et al. 2016 [147] FINRISK 2002 | Individuals (8101) from the FINRISK 2002 general population cohort (men and women aged 25 to 74 years). During a follow-up of 13 years, 813 subjects experienced an incident major adverse cardiovascular event, of which 116 were fatal. | Levels of C16:0, C18:0, and C24:1 and the ratios C18:0/C24:0, and C24:1/C24:0 were significantly higher in subjects with an incident major adverse cardiovascular event compared with asymptomatic subjects. C18:0 holds the potential for improving the risk classification over the Framingham risk score at a population level. C16:0 and C24:1 associated with recurrent major adverse cardiovascular event. |
| Wang et al. 2017 [148] PREDIMED | Participants (980) from the PREDIMED trial (Prevención con Dieta Mediterránea), including 230 incident cases of cardiovascular disease and 787 randomly selected participants at baseline. | Plasma concentrations of C16:0, C22:0, C24:0, and C24:1 were positively associated with incident cardiovascular disease risk. |
| Mantovani et al. 2018 [149] | Patients (581) with established or suspected coronary artery disease undergoing stress myocardial perfusion scintigraphy. | Higher plasmatic levels of C18:0, C20:0, C22:0, and C24:1 were associated with lower post stress anteroapical wall perfusion. Associations persisted after adjustment for conventional cardiovascular risk factors. |
| Anroedh et al. 2018 [150] ATHEROREMO | Patients (581) undergoing diagnostic coronary angiography or percutaneous coronary intervention for stable angina pectoris (SAP) or acute coronary syndrome (ACS). | C16:0 concentration was associated with major adverse cardiac events after adjustment for cardiac risk factors, clinical presentation, statin use, and HDL cholesterol level. After multivariable adjustment, concentrations of C16:0, C20:0, C24:1 and their ratios to C24:0 were associated with the composite endpoint death or nonfatal acute coronary syndrome. |
| Meeusen et al. 2018 [12] | Patients (504) between 18 and 75 years of age, who were undergoing clinically indicated coronary angiography. | Concentrations of C16:0, C18:0, and C24:1 and their ratios to C24:0 were significantly predictive for a combined outcome of myocardial infarction, coronary artery bypass graft, percutaneous intervention, stroke, and death at 4 years of follow-up. Concentrations of C16:0, C24:1 and the ratios C16:0/ C24:0, C18:0/ C24:0, and C24:1/C24:0 were significantly predictive for all-cause death at 18 years of follow-up and remained significant after adjusting for cardiovascular risk factors. |
| de Carvalho et al. 2018 [153] | Two cohorts of patients (337 and 119) with acute myocardial infarction, undergoing coronary angiography. | Identification of a 12-ceramide plasma prognostic signature that predicted long-term major adverse cardiac and cerebrovascular events in patients with acute myocardial infarction, of which C22:1, C24:1 and dihydro C16:0 were the strongest predictors. |
| Study | Population | Main Findings |
|---|---|---|
| Wigger et al. 2017 [158] | Individuals (288) from the D.E.S.I.R. cohort study (Data from Epidemiological Study on the Insulin Resistance syndrome) | The susceptibility to develop type 2 diabetes mellitus was associated with increased plasma levels of C18:0, C20:0, C22:0, and dihydro C22:0 |
| Lemaitre et al. 2018 [159] | Cohort of 2086 Native Americans without diabetes (average age of 38 years), 24% of whom had a BMI of 35 kg/m2 or greater. | Participants with higher (>90th percentile) plasmatic levels of C16:0, C18:0, C20:0, or C22:0 displayed hyperinsulinemia and insulin resistance |
| Study | Population | Main Findings |
|---|---|---|
| High altitude pulmonary edema (HAPE) | ||
| Guo et al. 2015 [168] | 22 HAPE subjects and 22 healthy controls | Plasma levels of C8-ceramide (and sphingosine) are increased in HAPE subjects compared to subjects not developing any symptoms after exposure to high altitude. |
| Bronchopulmonary dysplasia (BPD) | ||
| Laube et al. 2017 [171] van Mastrigt et al. 2018 [172] | 43 preterm infants (24–28 weeks): 25 received inhaled NO (iNO) and 18 placebo. 122 preterm babies (<32 weeks): 41 developed BPD. | aSMase activity in tracheal aspirates (TA) increases during the first two weeks of life and this increase is more pronounced in infants receiving iNOS. Ceramide levels are increased in TA following mechanical ventilation. Ceramide profiles changed over time with mechanical ventilation. Ceramide levels were lower in infants who developed BPD as compared to those who did not. |
| Congenital diaphragmatic hernia (CDH) and development of chronic lung disease | ||
| Snoek et al. 2016 [173] | 72 neonates (>34 weeks) with CHD. | Higher levels of ceramides (C18:1 and C24:0) in TA were found in patients in conventional mechanical ventilation compared to high-frequency oscillation. Levels of ceramides were not associated with a higher risk of mortality or chronic lung disease. |
| Pulmonary arterial hypertension (PAH) | ||
| Chen et al. 2014 [174] Brittain et al. 2016 [175] | 5 PAH patients and 5 control subjects. 19 PAH patients and 22 control subjects. | In lungs from PAH patients, levels of ceramide remains unchanged but levels of S-1P and its receptor S1PR2 are increased. Ceramides C16:0 and C24:0 were increased in right ventricle from PAH patients. |
| Cystic fibrosis (CF) | ||
| Guilbault et al. 2009 [176] Garic et al. 2017 [177] Brodlie et al. 2010 [178] | 58 patients with CF and 72 healthy control subjects. 15 CF patients and 15 health volunteers. 24 patients undergoing lung transplantation with CF (8), emphysema (8) or PH (8). | Plasma levels of ceramide were decreased in patients with CF. Plasma levels of C22:0, C24:0 and C26:0 were decreased in patients with CF whereas C16:0 was increased. Ceramide staining was increased in lungs from patients with CF or emphysema as compared to those from PAH patients or unused donors. C16:0, C18:0, and C20:0 (but not C22:0) were significantly increased in lungs with CF vs. PAH. |
| Chronic obstructive pulmonary disease (COPD) | ||
| Lea et al. 2016 [179] Scarpa et al. 2013 [180] Bowler et al. 2015 [181] | 10 patients with COPD and 7 controls (5 smokers). 35 patients with COPD and 11 nonsmoking controls. 250 patients with COPD, 122 of them with emphysema. | Levels of C16:0, C18:0 and C20:0 might be increased in alveolar macrophages from COPD patients. Lung ceramide was increased in COPD patients but was not related to the severity of the disease. Plasma ceramide levels were inversely correlated with emphysema severity whereas trihexosylceramide positively correlated with COPD exacerbations. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cogolludo, A.; Villamor, E.; Perez-Vizcaino, F.; Moreno, L. Ceramide and Regulation of Vascular Tone. Int. J. Mol. Sci. 2019, 20, 411. https://doi.org/10.3390/ijms20020411
Cogolludo A, Villamor E, Perez-Vizcaino F, Moreno L. Ceramide and Regulation of Vascular Tone. International Journal of Molecular Sciences. 2019; 20(2):411. https://doi.org/10.3390/ijms20020411
Chicago/Turabian StyleCogolludo, Angel, Eduardo Villamor, Francisco Perez-Vizcaino, and Laura Moreno. 2019. "Ceramide and Regulation of Vascular Tone" International Journal of Molecular Sciences 20, no. 2: 411. https://doi.org/10.3390/ijms20020411
APA StyleCogolludo, A., Villamor, E., Perez-Vizcaino, F., & Moreno, L. (2019). Ceramide and Regulation of Vascular Tone. International Journal of Molecular Sciences, 20(2), 411. https://doi.org/10.3390/ijms20020411