Murine Models of Acute Myeloid Leukaemia

Abstract
:1. Introduction
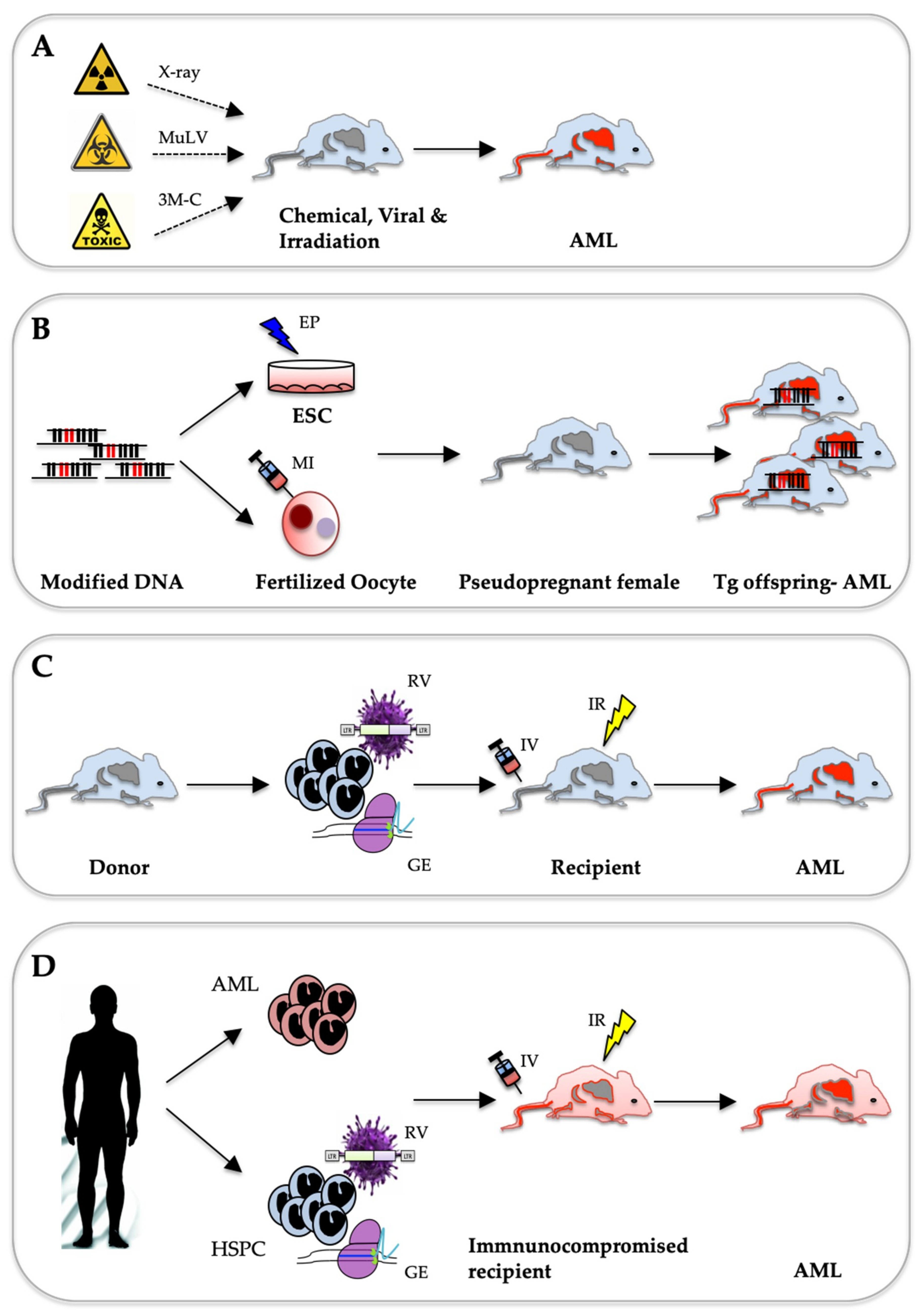
2. AML Mouse Models Induced by Chemicals, Viral Infection or Irradiation
2.1. Chemically Induced Leukaemia Models
2.2. Radiation-Induced Leukaemia Models
2.3. Virally Induced Leukaemia Models
3. Genetically Engineered Mouse Models
3.1. Conventional Transgenic AML Models
3.2. Transgenic AML Models by Homologous Recombination in ES Cells
3.3. Conditional Transgenic AML Mouse Models
3.3.1. Modelling AML-Associated Fusions
3.3.2. Modelling AML-Associated Mutations and Aberrantly Expressed Genes
4. Mouse Models Based on Adaptive Transfer of Hematopoietic Cells Virally Expressing an AML-Associated Proto-Oncogene
5. Modelling AML by Transferring Patient-Derived Cells into Immune-Compromised Mice
6. AML Mouse Models Generated with Genome-Editing Techniques
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.L.; Bailey, N.G. Acute Myeloid Leukemia Genetics: Risk Stratification and Implications for Therapy. Arch. Pathol. Lab. Med. 2015, 139, 1215–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. Chromosome translocations: Dangerous liaisons revisited. Nat. Rev. Cancer 2001, 1, 245–250. [Google Scholar] [CrossRef]
- Welch, J.; Ley, T.; Link, D.; Miller, C. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed] [Green Version]
- Meyer, S.C.; Levine, R.L. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol. 2014, 15, e382–e394. [Google Scholar] [CrossRef]
- Ito, S.; Barrett, A.J.; Dutra, A.; Pak, E.; Miner, S.; Keyvanfar, K.; Hensel, N.F.; Rezvani, K.; Muranski, P.; Liu, P.; et al. Long term maintenance of myeloid leukemic stem cells cultured with unrelated human mesenchymal stromal cells. Stem Cell Res. 2015, 14, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Alfred, L.J.; Wojdani, A.; Nieto, M.; Perez, R.; Yoshida, G. A chemical carcinogen, 3-methylcholanthrene, alters T-cell function and induces T-suppressor cells in a mouse model system. Immunology 1983, 207–212. [Google Scholar]
- Law, L.W.; Taormina, V.; Boyle, P.J. Response of Acute Lymphocytic Leukemias To the Purine Antagonist 6-Mercaptopurine. Ann. N. Y. Acad. Sci. 1954, 60, 244–250. [Google Scholar] [CrossRef]
- Skipper, H.E.; Perry, S. Kinetics Of Normal And Leukemic Leukocyte Populations And Relevance To Chemotherapy. Cancer Res. 1970, 30, 1883–1897. [Google Scholar] [PubMed]
- Chu, M.Y.; Fischer, G.A. A proposed mechanism of action of 1-β-d-arabinofuranosyl-cytosine as an inhibitor of the growth of leukemic cells. Biochem. Pharmacol. 1962, 11, 423–430. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Hirabayashi, Y.; Kaneko, T.; Kanno, J.; Kodama, Y.; Matsushima, Y.; Ogawa, Y.; Saitoh, M.; Sekita, K.; Uchida, O.; et al. Benzene-Induced Hematopoietic Neoplasms Including Myeloid Leukemia in Trp 53-Deficient C57BL/6 and C3H/He Mice. Toxicol. Sci. 2009, 110, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Robert, S. Leukemia and Benzene. Int. J. Environ. Res. Public Health 2012, 9, 2875–2893. [Google Scholar] [Green Version]
- Khalade, A.; Jaakkola, M.S.; Pukkala, E.; Jaakkola, J.J.K. Exposure to benzene at work and the risk of leukemia: A systematic review and meta-analysis. Environ. Heal. A Glob. Access Sci. Source 2010, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mchale, C.M.; Zhang, L.; Smith, M.T. Current understanding of the mechanism of benzene-induced leukemia in humans: Implications for risk assessment. Carcinogenesis 2012, 33, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, D.H.; Andersen, M.K.; Pedersen-Bjergaard, J. Mutations of AML1 are common in therapy-related myelodysplasia following therapy with alkylating agents and are significantly associated with deletion or loss of chromosome arm 7q and with subsequent leukemic transformation. Blood 2004, 104, 1474–1481. [Google Scholar] [CrossRef] [Green Version]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.T.; Wang, Y.H. Environmental and chemotherapeutic agents induce breakage at genes involved in leukemia-causing gene rearrangements in human hematopoietic stem/progenitor cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 779, 86–95. [Google Scholar] [CrossRef]
- Poynter, J.N.; Richardson, M.; Roesler, M.; Blair, C.K.; Hirsch, B.; Nguyen, P.; Cioc, A.; Cerhan, J.R.; Warlick, E. Chemical exposures and risk of acute myeloid leukemia and myelodysplastic syndromes in a population-based study. Int. J. Cancer 2017, 140. [Google Scholar] [CrossRef]
- Board, R.S.; Studies, L. Analysis of Cancer Risks in Populations Near Nuclear Facilities: Phase I; National Academy of Sciences: Washington, DC, USA, 2012. [Google Scholar]
- Finch, S.C. Radiation-induced leukemia: Lessons from history. Best Pract. Res. Clin. Haematol. 2007, 20, 109–118. [Google Scholar] [CrossRef]
- Balonov, M. Third annual Warren K. Sinclair keynote address: Retrospective analysis of impacts of the chernobyl accident. Health Phys. 2007, 93, 383–409. [Google Scholar] [CrossRef] [PubMed]
- Rivina, L.; Davoren, M.; Schiestl, R.H. Radiation-induced myeloid leukemia in murine models. Hum. Genom. 2014, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noshchenko, A.G.; Bondar, O.Y.; Drozdova, V.D. Radiation-induced leukemia among children aged 0–5 years at the time of the Chernobyl accident. Int. J. Cancer 2010, 127, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, F.; Bijwaard, H.; Bouffler, S.; Ellender, M.; Huiskamp, R.; Kowalczuk, C.; Meijne, E.; Sutmuller, M. A two-mutation model of radiation-induced acute myeloid leukemia using historical mouse data. Radiat. Environ. Biophys. 2011, 50, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Cadman, E.C.; Capizzi, R.L.; Bertino, J.R. Acute nonlymphocytic leukemia. A delayed complication of Hodgkin’s disease therapy: Analysis of 109 cases. Cancer 1977, 40, 1280–1296. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Belch, A.R.; Jacobs, A.; Bowen, D.; Padua, R.A.; Paietta, E.; Stanley, E.R. Increased circulating colony-stimulating factor-1 in patients with preleukemia, leukemia, and lymphoid malignancies. Blood 1991, 77, 1796–1803. [Google Scholar] [PubMed]
- Haran-Ghera, N.; Krautghamer, R.; Lapidot, T.; Peled, A.; Dominguez, M.G.; Stanley, E.R. Increased circulating colony-stimulating factor-1 (CSF-1) in SJL/J mice with radiation-induced acute myeloid leukemia (AML) is associated with autocrine regulation of AML cells by CSF-1. Blood 1997, 89, 2537–2545. [Google Scholar]
- Peng, Y.; Borak, T.B.; Bouffler, S.D.; Ullrich, R.L.; Weil, M.M.; Bedford, J.S. Radiation Leukemogenesis in Mice: Loss of PU.1 on Chromosome 2 in CBA and C57BL/6 Mice after Irradiation with 1 GeV/nucleon 56 Fe Ions, X Rays or γ Rays. Part I. Experimental Observation. Radiat. Res. 2009, 171, 484–493. [Google Scholar] [CrossRef]
- Olme, C.H.; Finnon, R.; Brown, N.; Kabacik, S.; Bouffler, S.D.; Badie, C. Live cell detection of chromosome 2 deletion and Sfpi1/PU1 loss in radiation-induced mouse acute myeloid leukaemia. Leuk. Res. 2013, 37, 1374–1382. [Google Scholar] [CrossRef] [Green Version]
- Verbiest, T.; Finnon, R.; Brown, N.; Cruz-Garcia, L.; Finnon, P.; O’Brien, G.; Ross, E.; Bouffler, S.; Scudamore, C.L.; Badie, C. Tracking preleukemic cells in vivo to reveal the sequence of molecular events in radiation leukemogenesis. Leukemia 2018, 32, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Klymenko, S.V.; Smida, J.; Atkinson, M.J.; Bebeshko, V.G.; Nathrath, M.; Rosemann, M. Allelic imbalances in radiation-associated acute myeloid leukemia. Genes 2011, 2, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jones, L.; Gaillard, C.; Binnewies, M.; Ochoa, R.; Garcia, E.; Lam, V.; Wei, G.; Yang, W.; Lobe, C.; et al. Initially disadvantaged, TEL-AML1 cells expand and initiate leukemia in response to irradiation and cooperating mutations. Leukemia 2013, 27, 1570–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rein, A. Murine leukemia viruses: Objects and organisms. Adv. Virol. 2011, 2011. [Google Scholar] [CrossRef]
- Friend, C. Cell-free transmission in adult swiss mice of a disease having the character of leukemia. J. Exp. Med. 1957, 105, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Largaespada, D.A. Genetic heterogeneity in acute myeloid leukemia: Maximizing information flow from MuLV mutagenesis studies. Leukemia 2000, 14, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Cmarik, J.; Ruscetti, S. Friend spleen focus-forming virus activates the tyrosine kinase sf-Stk and the transcription factor PU.1 to cause a multi-stage erythroleukemia in mice. Viruses 2010, 2, 2235–2257. [Google Scholar] [CrossRef]
- Moreau-Gachelin, F. Multi-stage Friend murine erythroleukemia: Molecular insights into oncogenic cooperation. Retrovirology 2008, 5, 1–11. [Google Scholar] [CrossRef]
- Singer, D.; Cooper, M.; Maniatis, G.M.; Marks, P.A.; Rifkind, R.A. Erythropoietic differentiation in colonies of cells transformed by Friend virus. Proc. Natl. Acad. Sci. USA. 1974, 71, 2668–2670. [Google Scholar] [CrossRef]
- Rao, G.; Rekhtman, N.; Cheng, G.; Krasikov, T.; Skoultchi, A.I. Deregulated expression of the PU.1 transcription factor blocks murine erythroleukemia cell terminal differentiation. Oncogene 1997, 14, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Kondoh, N.; Matsumoto, M.; Yoshida, M.; Maekawa, A.; Oikawa, T. Overexpression of PU.1 induces growth and differentiation inhibition and apoptotic cell death in murine erythroleukemia cells. Blood 1997, 89, 1383–1393. [Google Scholar]
- Mucenski, M.L.; Taylor, B.A.; Ihle, J.N.; Hartley, J.W.; Morse, H.C.; Jenkins, N.A.; Copeland, N.G. Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors. Mol. Cell. Biol. 1988, 8, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.; Koller, R.; Hu, X.; Anver, M.R. A Moloney murine leukemia virus-based retrovirus with 4070A long terminal repeat sequences induces a high incidence of myeloid as well as lymphoid neoplasms. J. Virol. 2003, 77, 4965–4971. [Google Scholar] [CrossRef] [PubMed]
- Kool, J.; Berns, A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat. Rev. Cancer 2009, 9, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Ranzani, M.; Annunziato, S.; Adams, D.J.; Montini, E. Cancer Gene Discovery: Exploiting Insertional Mutagenesis. Mol. Cancer Res. 2013, 11, 1141–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassiliou, G.S.; Cooper, J.L.; Rad, R.; Li, J.; Rice, S.; Uren, A.; Rad, L.; Ellis, P.; Andrews, R.; Banerjee, R.; et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 2011, 43, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Lauchle, J.O.; Kim, D.; Le, D.T.; Akagi, K.; Crone, M.; Krisman, K.; Warner, K.; Bonifas, J.M.; Li, Q.; Coakley, K.M.; et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature 2009, 461, 411–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.; Harris, W.; Pinkert, C.; Corcoran, L.; Alexander, W.; Cory, S.; Palmiter, R.; Brinster, R. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V.; Pattengale, P.K.; Weir, L.; Leder, P. Transgenic mice bearing the human c-myc gene activated by an immunoglobulin enhancer: A pre-B-cell lymphoma model. Proc. Natl. Acad. Sci. USA 1988, 85, 6047–6051. [Google Scholar] [CrossRef] [PubMed]
- Würtele, H.; Little, K.C.E.; Chartrand, P. Illegitimate DNA integration in mammalian cells. Gene Ther. 2003, 10, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Grisolano, J.L.; Wesselschmidt, R.L.; Pelicci, P.G.; Ley, T.J. Altered Myeloid Development and Acute Leukemia in Transgenic Mice Expressing PML-RARα Under Control of Cathepsin G Regulatory Sequences. Blood 1997, 89, 376–387. [Google Scholar]
- Early, E.; Moore, M.A.; Kakizuka, A.; Nason-Burchenal, K.; Martin, P.; Evans, R.M.; Dmitrovsky, E. Transgenic expression of PML/RARA impairs myelopoiesis. Proc. Natl. Acad. Sci. USA 1996, 93, 7900–7904. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Kogan, S.; Lagasse, E.; Weissman, I.; Alcalay, M.; Pelicci, P.G.; Atwater, S.; Bishop, J.M. A PMLRAR transgene initiates murine acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 2551–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Thé, H.; Chen, Z. Acute promyelocytic leukaemia: Novel insights into the mechanisms of cure. Nat. Rev. Cancer 2010, 10, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.X.; Zhu, X.H.; Men, X.Q.; Wang, L.; Huang, Q.H.; Jin, X.L.; Xiong, S.M.; Zhu, J.; Guo, W.M.; Chen, J.Q.; et al. Distinct leukemia phenotypes in transgenic mice and different corepressor interactions generated by promyelocytic leukemia variant fusion genes PLZF-RARA and NPM-RARA. Proc. Natl. Acad. Sci. USA 1999, 96, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Nasr, R.; Zhu, J.; Bazarbachi, A.; Lallemand-Breittenbach, V.; de Thé, H. How animal models of leukaemias have already benefited patients. Mol. Oncol. 2013, 7, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Grisendi, S.; Pandolfi, P.P. Modelling haematopoietic malignancies in the mouse and therapeutical implications. Oncogene 2002, 21, 3445–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, E.; Bruserud, O.; Gjertsen, B.T. Review: Genetic models of acute myeloid leukaemia. Oncogene 2008, 27, 3765–3779. [Google Scholar] [CrossRef]
- Tomasson, M.H.; Williams, I.R.; Hasserjian, R.; Udomsakdi, C.; McGrath, S.M.; Schwaller, J.; Druker, B.; Gilliland, D.G. TEL/PDGFbetaR induces hematologic malignancies in mice that respond to a specific tyrosine kinase inhibitor. Blood 1999, 93, 1707–1714. [Google Scholar]
- Rhoades, K.L.; Hetherington, C.J.; Harakawa, N.; Yergeau, D.A.; Zhou, L.; Liu, L.Q.; Little, M.T.; Tenen, D.G.; Zhang, D.E. Analysis of the role of AML1-ETO in leukemogenesis, using an inducible transgenic mouse model. Blood 2000, 96, 2108–2115. [Google Scholar]
- Yuan, Y.; Zhou, L.; Miyamoto, T.; Iwasaki, H.; Harakawa, N.; Hetherington, C.J.; Burel, S.A.; Lagasse, E.; Weissman, I.L.; Akashi, K.; et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 10398–10403. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.H.; Landrette, S.F.; Heilman, S.A.; Perrat, P.N.; Garrett, L.; Liu, P.P.; Le Beau, M.M.; Kogan, S.C.; Castilla, L.H. Cbfβ-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell 2006, 9, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Kumar, A.R.; Hudson, W.A.; Li, Q.; Wu, B.; Staggs, R.A.; Lund, E.A.; Sam, T.N.; Kersey, J.H. Malignant Transformation Initiated by Mll-AF9: Gene Dosage and Critical Target Cells. Cancer Cell 2008, 13, 432–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugale, A.; Norddahl, G.L.; Wahlestedt, M.; Säwén, P.; Jaako, P.; Pronk, C.J.; Soneji, S.; Cammenga, J.; Bryder, D. Hematopoietic Stem Cells Are Intrinsically Protected against MLL-ENL-Mediated Transformation. Cell Rep. 2014, 9, 1246–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulou, V.; Almosailleakh, M.; Royo, H.; Spetz, J.; Juge, S.; Brault, L.; Kopp, P.; Iacovino, M.; Kyba, M.; Tzankov, A.; et al. A Novel Inducible Mouse Model of MLL-ENL-driven Mixed-lineage Acute Leukemia. HemaSphere 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512. [Google Scholar] [CrossRef]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.J.; King, G.; Rabbitts, T.H. An MII-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-oliveira, M.S. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 273–284. [Google Scholar] [CrossRef]
- Collins, E.C.; Pannell, R.; Simpson, E.M.; Forster, A.; Rabbitts, T.H. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. Sci. Rep. 2000, 1, 127–132. [Google Scholar]
- Dobson, C.L.; Warren, A.J.; Pannell, R.; Forster, A.; Rabbitts, T.H. Tumorigenesis in mice with a fusion of the leukaemia oncogene MII and the bacterial lacZ gene. EMBO J. 2000, 19, 843–851. [Google Scholar] [CrossRef]
- Johnson, J.J.; Chen, W.; Hudson, W.; Yao, Q.; Taylor, M.; Rabbitts, T.H.; Kersey, J.H. Prenatal and postnatal myeloid cells demonstrate stepwise progression in the pathogenesis of MLL fusion gene leukemia. Blood 2003, 101, 3229–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayton, P.M.; Cleary, M.L. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Sinha, C.; Cunningham, L.C.; Liu, P.P. Core Binding Factor Acute Myeloid Leukemia: New Prognostic Categories and Therapeutic Opportunities. Semin. Hematol. 2015, 52, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, T.; Cai, Z.; Yang, S.; Lenny, N.; Lyu, C.J.; van Deursen, J.M.; Harada, H.; Downing, J.R. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood 1998, 91, 3134–3143. [Google Scholar] [PubMed]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R.; Bae, S.; Yamaguchi-Iwai, Y.; Ogawa, E.; Maruyama, M.; Inuzuka, M.; et al. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef]
- Speck, N.A.; Iruela-Arispe, M.L. Conditional Cre/LoxP strategies for the study of hematopoietic stem cell formation. Blood Cells Mol. Dis. 2009, 43, 6–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furth, P.A.; Onget, L.S.; B6gert, H.; Grusst, P.; Gossen, M.; Kistner, A.; Bujard, H.; Hennighausentl, L. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Biochemistry 1994, 91, 9302–9306. [Google Scholar] [CrossRef]
- Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.; Bujardt, H. Transcriptional Activation by Tetracyclines in Mammalian Cells. Science 1995, 268, 1766–1770. [Google Scholar] [CrossRef]
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.-J.; Downing, J.R. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Claij, N.; Wal, A. Van Der; Dekker, M. DNA Mismatch Repair Deficiency Stimulates N-Ethyl-N-nitrosourea-induced Mutagenesis and Lymphomagenesis. Cancer Res. 2003, 63, 2062–2066. [Google Scholar]
- Cabezas-Wallscheid, N.; Eichwald, V.; de Graaf, J.; Löwer, M.; Lehr, H.A.; Kreft, A.; Eshkind, L.; Hildebrandt, A.; Abassi, Y.; Heck, R.; et al. Instruction of haematopoietic lineage choices, evolution of transcriptional landscapes and cancer stem cell hierarchies derived from an AML1-ETO mouse model. EMBO Mol. Med. 2013, 5, 1804–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidenreich, O.; Riehle, H.; Hadwiger, P.; John, M.; Heil, G.; Vornlocher, H.; Nordheim, A. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t (8; 21)-positive leukemic cells. Blood 2003, 101, 3157–3163. [Google Scholar] [CrossRef] [PubMed]
- Nick, H.J.; Kim, H.; Chang, C.; Harris, K.W.; Reddy, V.; Klug, A.; Dc, W.; Klug, C.A. Distinct classes of c-Kit–activating mutations differ in their ability to promote RUNX1-ETO–associated acute myeloid leukemia. Blood 2012, 119, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Schessl, C.; Rawat, V.P.S.; Cusan, M.; Deshpande, A.; Kohl, T.M.; Rosten, P.M.; Spiekermann, K.; Humphries, R.K.; Schnittger, S.; Kern, W.; et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J. Clin. Investig. 2005, 115, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sportoletti, P.; Varasano, E.; Rossi, R.; Mupo, A.; Tiacci, E.; Vassiliou, G.; Martelli, M.P.; Falini, B. Mouse models of NPM1-mutated acute myeloid leukemia: Biological and clinical implications. Leukemia 2015, 29, 269–278. [Google Scholar] [CrossRef]
- Cheng, K.; Sportoletti, P.; Ito, K.; Clohessy, J.G.; Teruya-feldstein, J.; Kutok, J.L.; Pandolfi, P.P. Brief report The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood 2018, 115, 3341–3346. [Google Scholar] [CrossRef]
- Chou, S.H.; Ko, B.S.; Chiou, J.S.; Hsu, Y.C.; Tsai, M.H.; Chiu, Y.C.; Yu, I.S.; Lin, S.W.; Hou, H.A.; Kuo, Y.Y.; et al. A Knock-In Npm1 Mutation in Mice Results in Myeloproliferation and Implies a Perturbation in Hematopoietic Microenvironment. PLoS ONE 2012, 7, 1–10. [Google Scholar] [CrossRef]
- Mallardo, M.; Caronno, A.; Pruneri, G.; Raviele, P.R.; Viale, A.; Pelicci, P.G.; Colombo, E. NPMc+ and FLT3_ITD mutations cooperate in inducing acute leukaemia in a novel mouse model. Leukemia 2013, 27, 2248–2251. [Google Scholar] [CrossRef]
- Garg, M.; Nagata, Y.; Kanojia, D.; Mayakonda, A.; Yoshida, K.; Keloth, S.H.; Zang, Z.J.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood 2015, 126, 2491–2502. [Google Scholar] [CrossRef]
- Lee, B.H.; Tothova, Z.; Levine, R.L.; Anderson, K.; Buza-Vidas, N.; Cullen, D.E.E.; McDowell, E.P.; Adelsperger, J.; Fröhling, S.; Huntly, B.J.P.; et al. FLT3 Mutations Confer Enhanced Proliferation and Survival Properties to Multipotent Progenitors in a Murine Model of Chronic Myelomonocytic Leukemia. Cancer Cell 2007, 12, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Williams, I.R.; Anastasiadou, E.; Boulton, C.L.; Joseph, S.W.; Amaral, S.M.; Curley, D.P.; Duclos, N.; Huntly, B.J.P.; Fabbro, D.; et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene 2005, 24, 7882–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharazi, S.; Mead, A.J.; Mansour, A.; Hultquist, A.; Böiers, C.; Luc, S.; Buza-Vidas, N.; Ma, Z.; Ferry, H.; Atkinson, D.; et al. Impact of gene dosage, loss of wild-type allele, and FLT3 ligand on Flt3-ITD-induced myeloproliferation. Blood 2011, 118, 3613–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Bailey, E.; Greenblatt, S.; Huso, D.; Small, D. Loss of the wild-type allele contributes to myeloid expansion and disease aggressiveness in FLT3/ITD knockin mice. Blood 2011, 118, 4935–4945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovey, O.M.; Cooper, J.L.; Mupo, A.; Grove, C.S.; Lynn, C.; Conte, N.; Andrews, R.M.; Pacharne, S.; Tzelepis, K.; Vijayabaskar, M.S.; et al. Molecular synergy underlies the co-occurrence patterns and phenotype of NPM1-mutant acute myeloid leukemia. Blood 2017, 130, 1911–1922. [Google Scholar] [CrossRef]
- Mupo, A.; Celani, L.; Dovey, O.; Cooper, J.L.; Grove, C.; Rad, R.; Sportoletti, P.; Falini, B.; Bradley, A.; Vassiliou, G.S. A powerful molecular synergy between mutant Nucleophosmin and Flt3-ITD drives acute myeloid leukemia in mice. Leukemia 2013, 27, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Zorko, N.A.; Bernot, K.M.; Whitman, S.P.; Siebenaler, R.F.; Ahmed, E.H.; Marcucci, G.G.G.G.; Yanes, D.A.; McConnell, K.K.; Mao, C.; Kalu, C.; et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood 2012, 120, 1130–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, S.; Li, L.; Slape, C.; Nguyen, B.; Novak, R.; Duffield, A.; Huso, D.; Desiderio, S.; Borowitz, M.J.; Aplan, P.; et al. Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood 2012, 119, 2883–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annesley, C.E.; Rabik, C.; Duffield, A.S.; Rau, R.E.; Li, L.; Huff, V.; Small, D.; Loeb, D.M.; Brown, P. Knock-in of the Wt1 R394W mutation causes MDS and cooperates with Flt3/ITD to drive aggressive myeloid neoplasms in mice. Oncotarget 2018, 9, 35313–35326. [Google Scholar] [CrossRef]
- Meyer, S.E.; Qin, T.; Muench, D.E.; Masuda, K.; Venkatasubramanian, M.; Orr, E.; Suarez, L.; Gore, S.D.; Delwel, R.; Paietta, E.; et al. Dnmt3a haploinsufficiency transforms Flt3-ITD myeloproliferative disease into a rapid, spontaneous, and fully-penetrant acute myeloid leukemia. Cancer Discov. 2016, 6, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wang, E.; Zuber, J.; Rappaport, A.; Taylor, M.; Johns, C.; Lowe, S.W.; Vakoc, C.R. The Polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;Nras-G12D acute myeloid leukemia J. Oncogene 2013, 32, 930–938. [Google Scholar] [CrossRef]
- Omidvar, N.; Kogan, S.; Beurlet, S.; Le Pogam, C.; Janin, A.; West, R.; Noguera, M.E.; Reboul, M.; Soulie, A.; Leboeuf, C.; et al. BCL-2 and mutant NRAS interact physically and functionally in a mouse model of progressive myelodysplasia. Cancer Res. 2007, 67, 11657–11667. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Pulikkan, J.A.; Valk, P.J.M.; Castilla, L.H. Nras G12D oncoprotein inhibits apoptosis of preleukemic cells expressing Cbfb -SMMHC via activation of MEK/ERK axis. Blood 2014, 124, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.T.; Kutok, J.L.; Williams, I.R.; Cohen, S.; Moore, S.; Shigematsu, H.; Ley, T.J.; Akashi, K.; Beau, M.M. Le; Gilliland, D.G.; et al. Oncogenic K-ras cooperates with PML-RAR to induce an acute promyelocytic leukemia–like disease. Blood 2012, 108, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Hinai, A.A.; Valk, P.J.M. Review: Aberrant EVI1 expression in acute myeloid leukaemia. Br. J. Haematol. 2016, 172, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Suzuki, M.; Otsuki, A.; Shimizu, R.; Bresnick, E.H.; Engel, J.D.; Yamamoto, M. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 2014, 25, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; De Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; Van Der Velden, V.H.J.; Havermans, M.; Avellino, R.; Van Lom, K.; et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in Leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, E.; Wilson, M.P.; McGrath, K.E.; Li, A.J.; Frisch, B.J.; Palis, J.; Calvi, L.M.; Zhang, Y.; Perkins, A.S. EVI1 overexpression reprograms hematopoiesis via upregulation of Spi1 transcription. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Whitelaw, C.B.A.; Archibald, A.L.; Harris, S.; McClenaghan, M.; Simons, J.P.; Clark, A.J. Targeting expression to the mammary gland: Intronic sequences can enhance the efficiency of gene expression in transgenic mice. Transgenic Res. 1991, 1, 3–13. [Google Scholar] [CrossRef]
- Takai, J.; Moriguchi, T.; Suzuki, M.; Yu, L.; Ohneda, K.; Yamamoto, M. The Gata1 5’ region harbors distinct cis-regulatory modules that direct gene activation in erythroid cells and gene inactivation in HSCs. Blood 2013, 112, 3450–3460. [Google Scholar] [CrossRef]
- Daley, G.Q.; Van Etten, R.A.; Baltimore, D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Am. Assoc. Adv. Sci. 1990, 247, 824–830. [Google Scholar] [CrossRef]
- Mc Cormack, E.; Bruserud, Ø.; Gjertsen, B. Animal models of acute myelogenous leukaemia—Development, application and future perspectives. Leukemia 2005, 19, 687–706. [Google Scholar] [CrossRef] [PubMed]
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Huntly, B.J.P.; Shigematsu, H.; Deguchi, K.; Lee, B.H.; Mizuno, S.; Duclos, N.; Rowan, R.; Amaral, S.; Curley, D.; Williams, I.R.; et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004, 6, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Bindels, E.M.J.; Havermans, M.; Lugthart, S.; Erpelinck, C.; Wocjtowicz, E.; Krivtsov, A.V.; Rombouts, E.; Armstrong, S.A.; Taskesen, E.; Haanstra, J.R.; et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood 2012, 119, 5838–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavau, C.; Szilvassy, S.J.; Slany, R.; Cleary, M.L. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997, 16, 4226–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzio, A.; Passegué, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035. [Google Scholar] [CrossRef] [Green Version]
- de Guzman, C.G.; Warren, A.J.; Zhang, Z.; Gartland, L.; Erickson, P.; Drabkin, H.; Hiebert, S.W.; Klug, C.A. Hematopoietic Stem Cell Expansion and Distinct Myeloid Developmental Abnormalities in a Murine Model of the AML1-ETO Translocation. Mol. Cell. Biol. 2002, 22, 5506–5517. [Google Scholar] [CrossRef] [Green Version]
- So, C.W.; Karsunky, H.; Passegué, E.; Cozzio, A.; Weissman, I.L.; Cleary, M.L. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 2003, 3, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Heuser, M.; Yun, H.; Berg, T.; Yung, E.; Argiropoulos, B.; Kuchenbauer, F.; Park, G.; Hamwi, I.; Palmqvist, L.; Lai, C.K.; et al. Cell of Origin in AML: Susceptibility to MN1-Induced Transformation Is Regulated by the MEIS1/AbdB-like HOX Protein Complex. Cancer Cell 2011, 20, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.H.; Sadelain, M. The genetic engineering of hematopoietic stem cells: The rise of lentiviral vectors, the conundrum of the LTR, and the promise of lineage-restricted vectors. Mol. Ther. 2007, 15, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Victor Garcia-Martinez, J.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Theocharides, A.P.A.; Rongvaux, A.; Fritsch, K.; Flavell, R.A.; Manz, M.G. Humanized hemato-lymphoid system mice. Haematologica 2016, 101, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Nara, N.; Miyamoto, T. Direct and serial transplantation of human acute myeloid leukaemia into nude mice. Br. J. Cancer 1982, 45, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelebart, P.; Popa, M.; McCormack, E. Xenograft models of primary acute myeloid leukemia for the development of imaging strategies and evaluation of novel targeted therapies. Curr. Pharm. Biotechnol. 2016, 17, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L.; Gishizky, M.L.; Quan, S.; Golde, D.W.; Witte, O.N. Propagation of human blastic myeloid leukemias in the SCID mouse. Blood 1992, 79, 2089–2098. [Google Scholar] [PubMed]
- Cao, X.; Shores, E.W.; Hu-Li, J.; Anver, M.R.; Kelsail, B.L.; Russell, S.M.; Drago, J.; Noguchi, M.; Grinberg, A.; Bloom, E.T.; et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor γ chain. Immunity 1995, 2, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Ailles, L.E.; Gerhard, B.; Kawagoe, H.; Hogge, D.E. Growth characteristics of acute myelogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood 1999, 94, 1761–1772. [Google Scholar]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/ gamma c null mouse: An excellent recipient mouse model for engraftment of human cells. Bone 2002, 100, 3175–3182. [Google Scholar]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-scid IL2R null Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, M.; Chou, F.S.; Link, K.A.; Mizukawa, B.; Perry, R.L.; Carroll, M.; Mulloy, J.C. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 2010, 24, 1785–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billerbeck, E.; Barry, W.T.; Mu, K.; Dorner, M.; Rice, C.M.; Ploss, A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rγ(null) humanized mice. Blood 2011, 117, 3076–3086. [Google Scholar] [CrossRef]
- Feuring-Buske, M.; Gerhard, B.; Cashman, J.; Humphries, R.K.; Eaves, C.J.; Hogge, D.E. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia 2003, 17, 760–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.A.; Covassin, L.; Brehm, M.A.; Racki, W.; Pearson, T.; Leif, J.; Laning, J.; Fodor, W.; Foreman, O.; Burzenski, L.; et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 2009, 157, 104–118. [Google Scholar] [CrossRef]
- Covassin, L.; Laning, J.; Abdi, R.; Langevin, D.L.; Phillips, N.E.; Shultz, L.D.; Brehm, M.A. Human peripheral blood CD4 T cell-engrafted non-obese diabetic-scid IL2rgnull H2-Ab1tm1Gru Tg (human leucocyte antigen D-related 4) mice: A mouse model of human allogeneic graft-versus-host disease. Clin. Exp. Immunol. 2011, 166, 269–280. [Google Scholar] [CrossRef]
- Gopalakrishnapillai, A.; Kolb, E.A.; Dhanan, P.; Bojja, A.S.; Mason, R.W.; Corao, D.; Barwe, S.P. Generation of Pediatric Leukemia Xenograft Models in NSG-B2m Mice: Comparison with NOD/SCID Mice. Front. Oncol. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Cosgun, K.N.; Rahmig, S.; Mende, N.; Reinke, S.; Hauber, I.; Schäfer, C.; Petzold, A.; Weisbach, H.; Heidkamp, G.; Purbojo, A.; et al. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell 2014, 15, 227–238. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Mizutani, A.; Kawasaki, N.; Holmborn, K.; Sundqvist, A.; Tsutsumi, S.; Watabe, T.; Aburatani, H.; Heldin, C. Improved human erythropoiesis and platelet formation in humanized NSGW41 mice. Cell Rep. 2016, 6, 171–180. [Google Scholar]
- Culen, M.; Kosarova, Z.; Jeziskova, I.; Folta, A.; Chovancova, J.; Loja, T.; Tom, N.; Bystry, V.; Janeckova, V.; Dvorakova, D.; et al. The influence of mutational status and biological characteristics of acute myeloid leukemia on xenotransplantation outcomes in NOD SCID gamma mice. J. Cancer Res. Clin. Oncol. 2018, 144, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Dirnhofer, S.; Konantz, M.; Medinger, M.; Salih, H.R.; Rothfelder, K.; Tsakiris, D.A.; Passweg, J.R.; Lundberg, P.; Lengerke, C. Long-term observation reveals high-frequency engraftment of human acute myeloid leukemia in immunodeficient mice. Haematologica 2017, 102, 854–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griessinger, E.; Vargaftig, J.; Horswell, S.; Taussig, D.C.; Gribben, J.; Bonnet, D. Acute myeloid leukemia xenograft success prediction: Saving time. Exp. Hematol. 2018, 59, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Hogenesch, H.; Nikitin, A.Y. Challenges in pre-clinical testing of anti-cancer drugs in cell culture and in animal models. J. Control. Release 2012, 164, 183–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saland, E.; Boutzen, H.; Castellano, R.; Pouyet, L.; Griessinger, E.; Larrue, C.; De Toni, F.; Scotland, S.; David, M.; Danet-Desnoyers, G.; et al. A robust and rapid xenograft model to assess efficacy of chemotherapeutic agents for human acute myeloid leukemia. Blood Cancer J. 2015, 5, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.; Mizukawa, B.; Chou, F.S.; Sexton, C.; Shrestha, M.; Saunthararajah, Y.; Mulloy, J.C. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood 2013, 121, e90–e97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Abarrategi, A.; Mian, S.A.; Passaro, D.; Rouault-Pierre, K.; Grey, W.; Bonnet, D. Modeling the human bone marrow niche in mice: From host bone marrow engraftment to bioengineering approaches. J. Exp. Med. 2018, 215, 729–743. [Google Scholar] [CrossRef] [Green Version]
- Vaiselbuh, S.R.; Edelman, M.; Lipton, J.M.; Liu, J.M. Ectopic Human Mesenchymal Stem Cell-Coated Scaffolds in NOD/SCID Mice: An In Vivo Model of the Leukemia Niche. Tissue Eng. Part C Methods 2010, 16, 1523–1531. [Google Scholar] [CrossRef]
- Antonelli, A.; Noort, W.A.; Jaques, J.; de Boer, B.; de Jong-Korlaar, R.; Brouwers-Vos, A.Z.; Lubbers-Aalders, L.; van Velzen, J.F.; Bloem, A.C.; Schuringa, J.J. Establishing human leukemia xenograft mouse models by implanting human bone marrow–like scaffold-based niches. Blood 2016, 128, 2949–2960. [Google Scholar] [CrossRef]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Gentles, A.J.; Chatterjee, S.; Lan, F.; Reinisch, A.; Corces, M.R.; Xavy, S.; Shen, J.; Haag, D.; Chanda, S.; et al. Human AML-iPSCs Reacquire Leukemic Properties after Differentiation and Model Clonal Variation of Disease. Cell Stem Cell 2017, 20, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Kotini, A.G.; Chang, C.J.; Chow, A.; Yuan, H.; Ho, T.C.; Wang, T.; Vora, S.; Solovyov, A.; Husser, C.; Olszewska, M.; et al. Stage-Specific Human Induced Pluripotent Stem Cells Map the Progression of Myeloid Transformation to Transplantable Leukemia. Cell Stem Cell 2017, 20, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Kitano, A.; Jiang, Y.; Luu, V.; Hoegenauer, K.A.; Nakada, D. Clonal expansion and myeloid leukemia progression modeled by multiplex gene editing of murine hematopoietic progenitor cells. Exp. Hematol. 2018, 64, 33–44. [Google Scholar] [CrossRef]
- Tothova, Z.; Krill-Burger, J.M.; Popova, K.D.; Landers, C.C.; Sievers, Q.L.; Yudovich, D.; Belizaire, R.; Aster, J.C.; Morgan, E.A.; Tsherniak, A.; et al. Multiplex CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell 2017, 21, 547–555. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef]
- Breese, E.H.; Buechele, C.; Dawson, C.; Cleary, M.L.; Porteus, M.H. Use of genome engineering to create patient specific MLL translocations in primary human hematopoietic stem and progenitor cells. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef]
- Schneidawind, C.; Jeong, J.; Schneidawind, D.; Kim, I.-S.; Duque-Afonso, J.; Wong, S.H.K.; Iwasaki, M.; Breese, E.H.; Zehnder, J.L.; Porteus, M.; et al. MLL leukemia induction by t(9;11) chromosomal translocation in human hematopoietic stem cells using genome editing. Blood Adv. 2018, 2, 832–845. [Google Scholar] [CrossRef]
- Reimer, J.; Knöß, S.; Labuhn, M.; Charpentier, E.M.; Göhring, G.; Schlegelberger, B.; Klusmann, J.H.; Heckl, D. CRISPR-Cas9-induced t(11;19)/MLL-ENL translocations initiate leukemia in human hematopoietic progenitor cells in vivo. Haematologica 2017, 102, 1558–1566. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Year | Transgene | Strategy | Promoter | Inducer | Cellular Target | Phenotype | Ref. |
|---|---|---|---|---|---|---|---|
| 1996 | PML-RARA | Conventional | CD11b | Myeloid lineage (BM, periphery) | Abnormal myelopoiesis. No APL | [52] | |
| 1997 | PML-RARA | Conventional | hCG | Myeloid lineage (BM, periphery) | Myeloid cells expansion in BM and spleen. AML-like with 30% penetrance after long (> 100 days) latency | [51] | |
| 1997 | PML-RARA | Conventional | hMRP8 | Myeloid lineage (BM, periphery) | APL-like disease (median 174 days) | [53] | |
| 2000 | RUNX1-ETO | Conditional | Tet | tTA | BM | Abnormal haematopoiesis. No AML | [60] |
| 2001 | RUNX1-ETO | Conventional | hMRP8 | Myeloid (neutrophils & monocytes) | AML-Only upon new-born treatment with ENU | [61] | |
| 2006 | Cbfb-MYH11 | Conditional | Cbfβ | Mx-iCre | BM (LSK) | AML-Aberrant myeloid progenitors, blocked megakaryotic differentiation. | [62] |
| 2008 | Mll-AF9 | Knock-in (Mll1; Mllex8-AF9 cDNA) | Mll | AML-Higher MLL-AF9 expression in HSCs than GMPs. | [63] | ||
| 2014 | MLL-ENL | Conditional | TRE (Col1a) | rtTA | LT-HS, pMeg/E, HSC, MPP, GMLP, CLP | AML- no leukaemia from HSC | [64] |
| 2016 | MLL-AF9 | Conditional | TRE (Hprt) | rtTA | LT-HSC, ST-HSC, CMP, GMP | AML-dependent on DOX dose and cellular origin | [65] |
| 2018 | MLL-ENL | Conditional | TRE (Hprt) | rtTA | LT-HSC, LMPP, CMP | AML-MLL-dependent on DOX dose and cellular target | [66] |
| Year | Co-Op Mutations | Activity | Promoter | Inducer | Cellular Target | Phenotype | Ref. |
|---|---|---|---|---|---|---|---|
| 2007 | NRAS12D + BCL2 | Const. | hMPP8 | Myeloid lineage (BM, periphery) | MDS/AML | [102] | |
| Cond. | Tet | rtTA | |||||
| 2012 | MLL-PTD + FLT3-ITD | Const. | Mll + Flt3 | Mll and Flt3 expressing cells | AML with 100% penetrance | [97] | |
| 2012 | NUP98-HOXD13 + FLT3-ITD | Conv. (FLT3-ITD) | Flt3 | Hematopoietic lineage cells (FL, BM) | AML with 100% penetrance | [98] | |
| Conv. (NUP98-HOXD12) | Vav | ||||||
| 2012 | KRAS-G12D + PML-RARA | Cond. (Kras-G12D) | Mx-iCre | Myeloid lineage (BM, periphery) | APL-like Disease with 69% penetrance, remaining mice developed MDS | [104] | |
| Const. (PML-RARA) | hCG | ||||||
| 2013 | NPM1c + FLT3-ITD | Cond. (NPM1c) | Mx1 | Mx-iCre | Hematopoietic lineage cells (BM) | AML after short latency (median 49 days) | [96] |
| Const. (Flt3-ITD) | |||||||
| 2014 | NRAS-G12D + CBFβ-SMMHC | Cond. | Mx1 | Mx-iCre | Hematopoietic lineage cells (BM) | AML after short latency (median 13.7 weeks) and full penetrance | [103] |
| 2017 | NPM1c + NRAS-G12D | Cond. | Mx1 | Mx.iCre | Hematopoietic lineage cells (BM) | AML with 95% penetrance, some mice develop MPN | [46] |
| NPM1c + FLT3-ITD | AML with 100% penetrance | ||||||
| 2018 | WT1-R394W + FLT3-ITD | Const. | Wt1 and Flt3 | Wt1 and Flt3 expressing cells | MPN-like disease or T-ALL after short latency-AML associated with LOH of Flt3 | [99] |
| Year | Transgene | Viral Vector | Cellular Target | Phenotype | Ref. |
|---|---|---|---|---|---|
| 1990 | BCR-ABL | pMSCV-pgk-neo | Total BM | Myeloproliferative malignancy, ALL and CML-like | [111] |
| 1997 | MLL-ENL | pMSCV-IRES-GFP | Thy-1loSca-1+Hi-2Khi, 5-FU treated BM | Self-renewal in vitro & AML in vivo | [117] |
| 2002 | RUNX1-ETO | pMSCV-IRES-GFP | HSC c-Kit+Sca-1−Lin− | Myeloid developmental abnormality but no AML | [118] |
| 2003 | MLL-GAS7 | pMSCV-pgk-neo | HSPC | Mixed lineage leukaemia phenotype | [120] |
| 2004 | MOZ-TIF2, BCR-ABL | pMSCV-IRES-GFP | CMP, GMP | MOZ-TIF2 but not BCR-ABL resulted in transplantable AML in vivo | [114] |
| 2006 | MLL-AF9 | pMSCV-IRES-GFP | GMP | Transplantation of transduced cells propagated in MC resulted in AML in vivo | [115] |
| 2011 | MN1 | pMSCV-pgk-neo | CMP, GMP | CMP are susceptible for MN1 transformation, GMP required co-expression of MEIS1 for AML induction | [121] |
| 2012 | MLL-AF9 | pMSCV-pgk-puro | Evi1+/− MLL-AF9 transduced cells | Knockdown of Evi1 delayed leukaemia induction in vivo | [116] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almosailleakh, M.; Schwaller, J. Murine Models of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 453. https://doi.org/10.3390/ijms20020453
Almosailleakh M, Schwaller J. Murine Models of Acute Myeloid Leukaemia. International Journal of Molecular Sciences. 2019; 20(2):453. https://doi.org/10.3390/ijms20020453
Chicago/Turabian StyleAlmosailleakh, Marwa, and Juerg Schwaller. 2019. "Murine Models of Acute Myeloid Leukaemia" International Journal of Molecular Sciences 20, no. 2: 453. https://doi.org/10.3390/ijms20020453
APA StyleAlmosailleakh, M., & Schwaller, J. (2019). Murine Models of Acute Myeloid Leukaemia. International Journal of Molecular Sciences, 20(2), 453. https://doi.org/10.3390/ijms20020453