Microbial Metabolites Determine Host Health and the Status of Some Diseases


Abstract
:1. Introduction
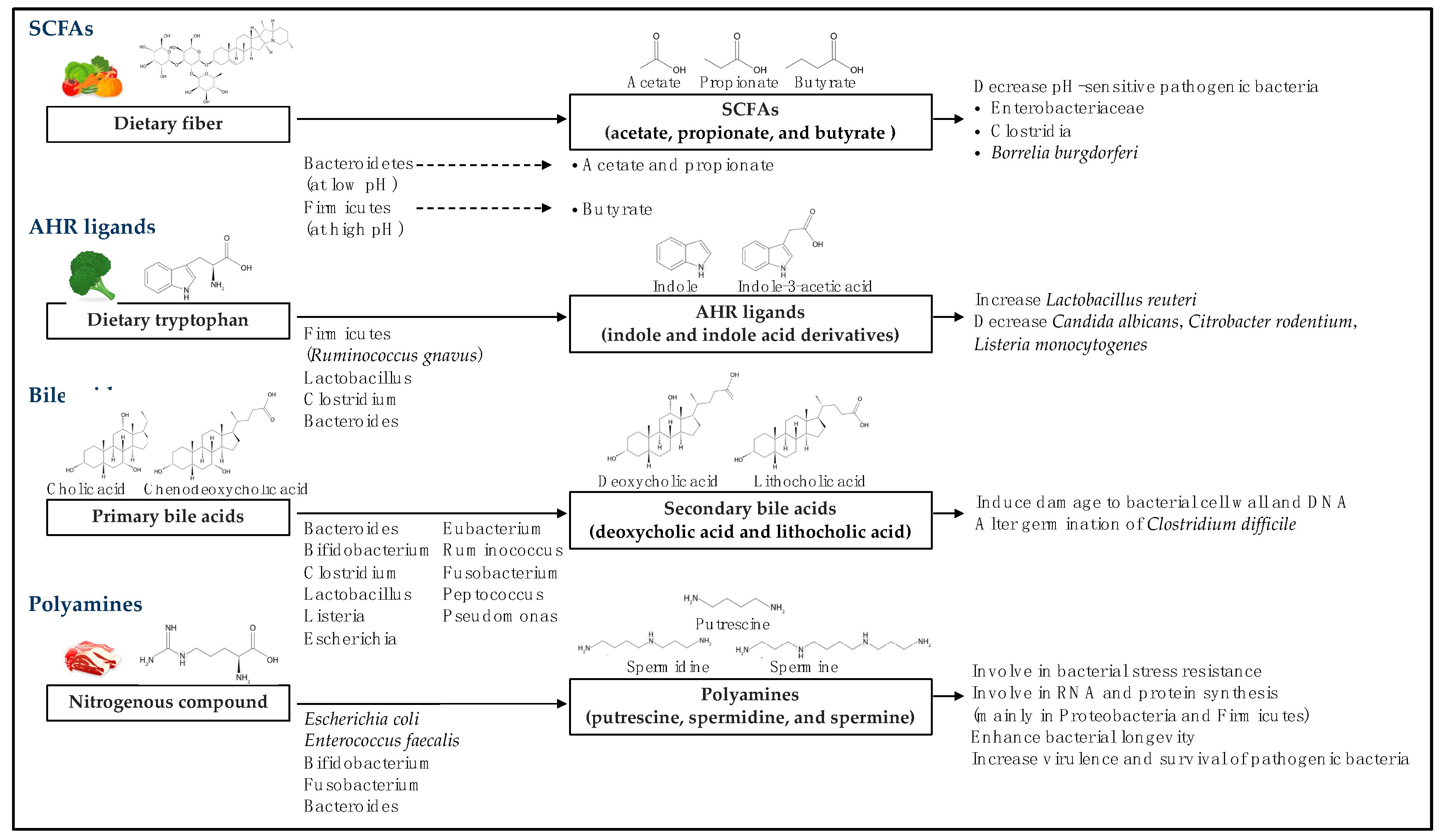
2. Production of Microbial Metabolites and Their Effect on Microbial Clades
2.1. Short-Chain Fatty Acids
2.2. Aryl Hydrocarbon Receptor Ligands
2.3. Bile Acids
2.4. Polyamines
2.5. Others
2.5.1. Equol
2.5.2. Compound K
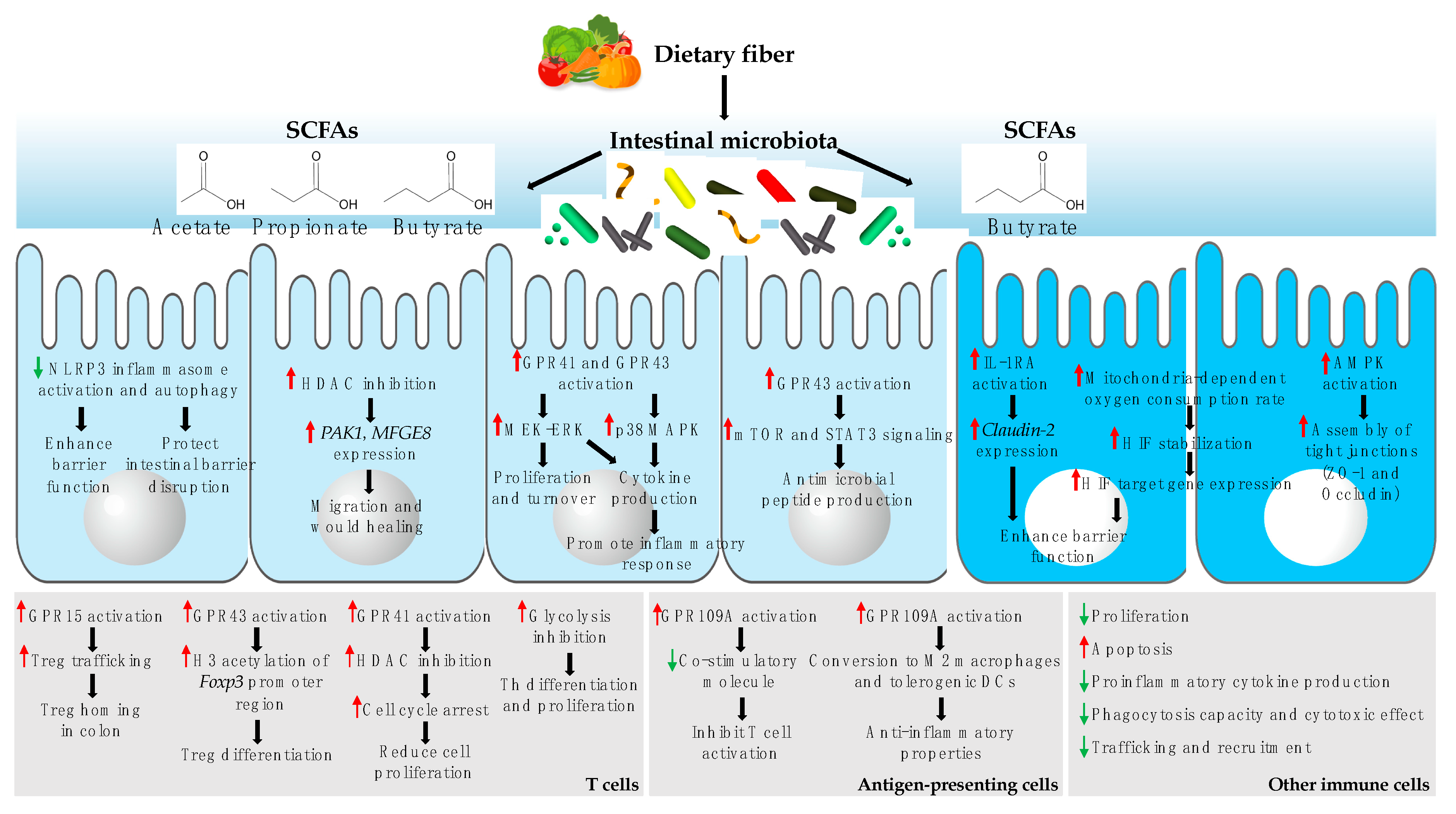
3. Microbial Metabolites: Messages from the Intestinal Microbiota to the Host Cell
3.1. Effect of SCFAs on Host Cells
3.1.1. Effect on Intestinal Epithelial Cells (IECs)
3.1.2. Effect on Immune Cells
3.2. Effect of AHR Ligands on Host Cells
3.2.1. Effect on IECs
3.2.2. Effect on Immune Cells
3.3. Effect of Bile Acids on Host Cells
3.3.1. Effect on IECs
3.3.2. Effect on Immune Cells
3.4. Effect of Polyamines on Host Cells
3.4.1. Effect on IECs
3.4.2. Effect on Immune Cells
3.5. Effect of Other Metabolites on Host Cells
3.5.1. Effect on IECs
3.5.2. Effect on Immune Cells
4. Metabolites and Diseases
4.1. IBDs
4.2. NAFLD
4.3. Obesity
4.4. Metabolic Diseases
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHR | aryl hydrocarbon receptor |
| BMDM | bone marrow-derived macrophage |
| cAMP | cyclic adenosine monophosphate |
| CBP | CREB-binding protein |
| CNS | central nervous system |
| CREB | cAMP response element binding protein |
| CVD | cardiovascular disease |
| DC | dendritic cell |
| DCA | deoxycholic acid |
| ERK | extracellular signal-regulated kinases |
| FXR | farnesoid X receptor |
| GI | gastrointestinal |
| GLP-1 | glucagon-like peptide-1 |
| GPR | G-protein coupled receptor |
| HDAC | histone deacetylase |
| I3C | indole-3-carbinol |
| IBD | inflammatory bowel disease |
| IEC | intestinal epithelial cell |
| IEL | intraepithelial lymphocyte |
| IL | interleukin |
| ILC | innate lymphoid cell |
| ILC3 | group 3 of innate lymphoid cell |
| LCA | lithocholic acid |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MEK | mitogen-activated protein kinase |
| MFGE8 | milk fat globule-EGF factor 8 |
| mTOR | mammalian target of rapamycin |
| NAFLD | non-alcoholic fatty liver disease |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP | NOD-like receptor pyrin domain-containing protein |
| PAKI | p21 activated kinase |
| ROS | reactive oxygen species |
| SCFA | short-chain fatty acid |
| SGLT1 | Na(+)/glucose co-transporter 1 |
| STAT | signal transducer and activator of transcription |
| T2D | type 2 diabetes |
| TGR | G-protein-coupled receptor |
| Th | helper T cell |
| TNF | tumor necrosis factor |
| Treg | regulatory T cell |
| VDR | vitamin D receptor |
References
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Hillman, E.T.; Lu, H.; Yao, T.; Nakatsu, C.H. Microbial Ecology along the Gastrointestinal Tract. Microbes Environ. 2017, 32, 300–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sittipo, P.; Lobionda, S.; Lee, Y.K.; Maynard, C.L. Intestinal microbiota and the immune system in metabolic diseases. J. Microbiol. 2018, 56, 154–162. [Google Scholar] [CrossRef]
- Brune, A.; Friedrich, M. Microecology of the termite gut: Structure and function on a microscale. Curr. Opin. Microbiol. 2000, 3, 263–269. [Google Scholar] [CrossRef]
- Gu, S.; Chen, D.; Zhang, J.N.; Lv, X.; Wang, K.; Duan, L.P.; Nie, Y.; Wu, X.L. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE 2013, 8, e74957. [Google Scholar] [CrossRef]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australas J. Anim. Sci. 2017, 30, 100–110. [Google Scholar] [CrossRef]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Elinav, E. Metabolites: Messengers between the microbiota and the immune system. Genes Dev. 2016, 30, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xie, G.; Zhao, A.; Zhao, L.; Yao, C.; Chiu, N.H.; Zhou, Z.; Bao, Y.; Jia, W.; Nicholson, J.K.; et al. The footprints of gut microbial-mammalian co-metabolism. J. Proteome Res. 2011, 10, 5512–5522. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Chen, Y.; Smith, T.C., 2nd; Karna, S.L.R.; Seshu, J. Short-Chain Fatty Acids Alter Metabolic and Virulence Attributes of Borrelia burgdorferi. Infect. Immun. 2018, 86, e00217-18. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Griffith, J.A.; Chasan-Taber, L.; Olendzki, B.C.; Jackson, E.; Stanek, E.J., 3rd; Li, W.; Pagoto, S.L.; Hafner, A.R.; Ockene, I.S. Association between dietary fiber and serum C-reactive protein. Am. J. Clin. Nutr. 2006, 83, 760–766. [Google Scholar] [CrossRef]
- Feng, W.; Ao, H.; Peng, C. Gut Microbiota, Short-Chain Fatty Acids, and Herbal Medicines. Front. Pharm. 2018, 9, 1354. [Google Scholar] [CrossRef]
- Postler, T.S.; Ghosh, S. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab. 2017, 26, 110–130. [Google Scholar] [CrossRef] [Green Version]
- Uchimura, Y.; Fuhrer, T.; Li, H.; Lawson, M.A.; Zimmermann, M.; Yilmaz, B.; Zindel, J.; Ronchi, F.; Sorribas, M.; Hapfelmeier, S.; et al. Antibodies Set Boundaries Limiting Microbial Metabolite Penetration and the Resultant Mammalian Host Response. Immunity 2018, 49, 545–559. [Google Scholar] [CrossRef]
- Parker, A.; Lawson, M.A.E.; Vaux, L.; Pin, C. Host-microbe interaction in the gastrointestinal tract. Environ. Microbiol. 2018, 20, 2337–2353. [Google Scholar] [CrossRef]
- Shibata, N.; Kunisawa, J.; Kiyono, H. Dietary and Microbial Metabolites in the Regulation of Host Immunity. Front. Microbiol. 2017, 8, 2171. [Google Scholar] [CrossRef]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef]
- Cummings, J.H. Dietary fibre. Br. Med. Bull. 1981, 37, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Mariotti, F.; Huneau, J.F.; Tome, D. Effects of amino acid-derived luminal metabolites on the colonic epithelium and physiopathological consequences. Amino Acids 2007, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Geypens, B.; Claus, D.; Evenepoel, P.; Hiele, M.; Maes, B.; Peeters, M.; Rutgeerts, P.; Ghoos, Y. Influence of dietary protein supplements on the formation of bacterial metabolites in the colon. Gut 1997, 41, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Gibson, G.R.; Beatty, E.; Cummings, J.H. Estimation of short-chain fatty acid production from protein by human intestinal bacteria based on branched-chain fatty acid measurements. FEMS Microbiol. Lett. 1992, 101, 81–88. [Google Scholar] [CrossRef]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.; Redondo-Blanco, S.; Gutiérrez-del-Río, I.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Colon microbiota fermentation of dietary prebiotics towards short-chain fatty acids and their roles as anti-inflammatory and antitumour agents: A review. J. Funct. Foods 2016, 25, 511–522. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Cherrington, C.A.; Hinton, M.; Pearson, G.R.; Chopra, I. Short-chain organic acids at ph 5.0 kill Escherichia coli and Salmonella spp. without causing membrane perturbation. J. Appl. Bacteriol. 1991, 70, 161–165. [Google Scholar] [CrossRef]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Prohaszka, L.; Jayarao, B.M.; Fabian, A.; Kovacs, S. The role of intestinal volatile fatty acids in the Salmonella shedding of pigs. Zent. Vet. B 1990, 37, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Rannug, A.; Stockinger, B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009, 30, 447–454. [Google Scholar] [CrossRef]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef]
- Murray, I.A.; Perdew, G.H. Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr. Opin. Toxicol. 2017, 2, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Ashida, H.; Fukuda, I.; Yamashita, T.; Kanazawa, K. Flavones and flavonols at dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS Lett. 2000, 476, 213–217. [Google Scholar] [CrossRef]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharm. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Shi, L.Z.; Faith, N.G.; Nakayama, Y.; Suresh, M.; Steinberg, H.; Czuprynski, C.J. The aryl hydrocarbon receptor is required for optimal resistance to Listeria monocytogenes infection in mice. J. Immunol. 2007, 179, 6952–6962. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.-m.E.; Fish, K.; Fu, Y.-X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Gérard, P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 2013, 3, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midtvedt, T. Microbial bile acid transformation. Am. J. Clin. Nutr. 1974, 27, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Merritt, M.E.; Donaldson, J.R. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. J. Med. Microbiol. 2009, 58 Pt 12, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Prieto, A.I.; Ramos-Morales, F.; Casadesus, J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics 2006, 174, 575–584. [Google Scholar] [CrossRef]
- Kandell, R.L.; Bernstein, C. Bile salt/acid induction of DNA damage in bacterial and mammalian cells: Implications for colon cancer. Nutr. Cancer 1991, 16, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Slocum, M.M.; Sittig, K.M.; Specian, R.D.; Deitch, E.A. Absence of intestinal bile promotes bacterial translocation. Am. Surg. 1992, 58, 305–310. [Google Scholar] [PubMed]
- Schäffler, H.; Breitrück, A. Clostridium difficile—From Colonization to Infection. Front. Microbiol. 2018, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Sorg, J.A.; Sonenshein, A.L. Inhibiting the Initiation of Clostridium difficile Spore Germination using Analogs of Chenodeoxycholic Acid, a Bile Acid. J. Bacteriol. 2010, 192, 4983. [Google Scholar] [CrossRef] [PubMed]
- Tofalo, R.; Cocchi, S.; Suzzi, G. Polyamines and Gut Microbiota. Front. Nutr. 2019, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Uda, K.; Tsujikawa, T.; Fujiyama, Y.; Bamba, T. Rapid absorption of luminal polyamines in a rat small intestine ex vivo model. J. Gastroenterol. Hepatol. 2003, 18, 554–559. [Google Scholar] [CrossRef]
- Matsumoto, M.; Benno, Y. The Relationship between Microbiota and Polyamine Concentration in the Human Intestine: A Pilot Study. Microbiol. Immunol. 2007, 51, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Kibe, R.; Ooga, T.; Aiba, Y.; Kurihara, S.; Sawaki, E.; Koga, Y.; Benno, Y. Impact of Intestinal Microbiota on Intestinal Luminal Metabolome. Sci. Rep. 2012, 2, 233. [Google Scholar] [CrossRef] [Green Version]
- Noack, J.; Dongowski, G.; Hartmann, L.; Blaut, M. The human gut bacteria Bacteroides thetaiotaomicron and Fusobacterium varium produce putrescine and spermidine in cecum of pectin-fed gnotobiotic rats. J. Nutr. 2000, 130, 1225–1231. [Google Scholar] [CrossRef]
- Gong, S.; Richard, H.; Foster, J.W. YjdE (AdiC) is the arginine:agmatine antiporter essential for arginine-dependent acid resistance in Escherichia coli. J. Bacteriol. 2003, 185, 4402–4409. [Google Scholar] [CrossRef]
- Suarez, C.; Espariz, M.; Blancato, V.S.; Magni, C. Expression of the agmatine deiminase pathway in Enterococcus faecalis is activated by the AguR regulator and repressed by CcpA and PTS(Man) systems. PLoS ONE 2013, 8, e76170. [Google Scholar] [CrossRef] [PubMed]
- Kitada, Y.; Muramatsu, K.; Toju, H.; Kibe, R.; Benno, Y.; Kurihara, S.; Matsumoto, M. Bioactive polyamine production by a novel hybrid system comprising multiple indigenous gut bacterial strategies. Sci. Adv. 2018, 4, eaat0062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, Y.; Nara, M.; Sakanaka, M.; Gotoh, A.; Kitakata, A.; Okuda, S.; Kurihara, S. Comprehensive analysis of polyamine transport and biosynthesis in the dominant human gut bacteria: Potential presence of novel polyamine metabolism and transport genes. Int. J. Biochem. Cell Biol. 2017, 93, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, M.; Sugiyama, Y.; Kitakata, A.; Katayama, T.; Kurihara, S. Carboxyspermidine decarboxylase of the prominent intestinal microbiota species Bacteroides thetaiotaomicron is required for spermidine biosynthesis and contributes to normal growth. Amino Acids 2016, 48, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.K.; Tabor, C.W.; Tabor, H. Polyamines protect Escherichia coli cells from the toxic effect of oxygen. Proc. Natl. Acad. Sci. USA 2003, 100, 2261–2265. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, G.; Rather, P.N. Evidence that putrescine acts as an extracellular signal required for swarming in Proteus mirabilis. Mol. Microbiol. 2004, 51, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305. [Google Scholar] [CrossRef]
- Jelsbak, L.; Thomsen, L.E.; Wallrodt, I.; Jensen, P.R.; Olsen, J.E. Polyamines Are Required for Virulence in Salmonella enterica Serovar Typhimurium. PLoS ONE 2012, 7, e36149. [Google Scholar] [CrossRef]
- Ware, D.; Watt, J.; Swiatlo, E. Utilization of putrescine by Streptococcus pneumoniae during growth in choline-limited medium. J. Microbiol. 2005, 43, 398–405. [Google Scholar]
- Wortham, B.W.; Oliveira, M.A.; Fetherston, J.D.; Perry, R.D. Polyamines are required for the expression of key Hms proteins important for Yersinia pestis biofilm formation. Environ. Microbiol. 2010, 12, 2034–2047. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, M.W.; Parker, Z.M.; Walter, N.E.; Rutkovsky, A.C.; Cartaya-Marin, C.; Karatan, E. Spermidine regulates Vibrio cholerae biofilm formation via transport and signaling pathways. FEMS Microbiol. Lett. 2009, 299, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Axelson, M.; Setchell, K.D. The excretion of lignans in rats—Evidence for an intestinal bacterial source for this new group of compounds. FEBS Lett. 1981, 123, 337–342. [Google Scholar] [CrossRef]
- Setchell, K.D.; Zimmer-Nechemias, L.; Cai, J.; Heubi, J.E. Isoflavone content of infant formulas and the metabolic fate of these phytoestrogens in early life. Am. J. Clin. Nutr. 1998, 68 (Suppl. 6), 1453s–1461s. [Google Scholar] [CrossRef]
- Setchell, K.D.R.; Clerici, C. Equol: History, chemistry, and formation. J. Nutr. 2010, 140, 1355S–1362S. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Roncaglia, L.; De Lucia, M.; Amaretti, A.; Leonardi, A.; Pagnoni, U.M.; Rossi, M. Bioconversion of soy isoflavones daidzin and daidzein by Bifidobacterium strains. Appl. Microbiol. Biotechnol. 2009, 81, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Rafii, F. The role of colonic bacteria in the metabolism of the natural isoflavone daidzin to equol. Metabolites 2015, 5, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kimura, S.; Ishii, Y.; Tateda, K. Equol inhibits growth and spore formation of Clostridioides difficile. J. Appl. Microbiol. 2019, 127, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, L.; Flórez, A.B.; Guadamuro, L.; Mayo, B. Effect of Soy Isoflavones on Growth of Representative Bacterial Species from the Human Gut. Nutrients 2017, 9, 727. [Google Scholar] [CrossRef]
- Hasebe, T.; Ueno, N.; Musch, M.W.; Nadimpalli, A.; Kaneko, A.; Kaifuchi, N.; Watanabe, J.; Yamamoto, M.; Kono, T.; Inaba, Y.; et al. Daikenchuto (TU-100) shapes gut microbiota architecture and increases the production of ginsenoside metabolite compound K. Pharmacol. Res. Perspect. 2016, 4, e00215. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, W. An Insight into Ginsenoside Metabolite Compound K as a Potential Tool for Skin Disorder. Evid.-Based Complement. Altern. Med. ECAM 2018, 2018, 8075870. [Google Scholar] [CrossRef]
- An, K.; Shengjie, Z.; Jinjun, S.; Liuqing, D. Gut microbiota-mediated deglycosylation of ginsenoside Rb1 in rats: In vitro and in vivo insights from quantitative ultra-performance liquid chromatography-mass spectrometry analysis. Anal. Methods 2015, 7, 6173–6181. [Google Scholar] [CrossRef]
- Leung, K.W.; Wong, A.S.-T. Pharmacology of ginsenosides: A literature review. Chin. Med. 2010, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, C.; Hasegawa, H.; Murata, J.; Saiki, I. In vivo antimetastatic action of ginseng protopanaxadiol saponins is based on their intestinal bacterial metabolites after oral administration. Oncol. Res. 1997, 9, 411–417. [Google Scholar] [PubMed]
- Van den Heuvel, E.G.; Wils, D.; Pasman, W.J.; Saniez, M.H.; Kardinaal, A.F. Dietary supplementation of different doses of NUTRIOSE FB, a fermentable dextrin, alters the activity of faecal enzymes in healthy men. Eur. J. Nutr. 2005, 44, 445–451. [Google Scholar] [CrossRef]
- Kim, K.-A.; Yoo, H.H.; Gu, W.; Yu, D.-H.; Jin, M.J.; Choi, H.-L.; Yuan, K.; Guerin-Deremaux, L.; Kim, D.-H. A prebiotic fiber increases the formation and subsequent absorption of compound K following oral administration of ginseng in rats. J. Ginseng Res. 2015, 39, 183–187. [Google Scholar] [CrossRef]
- Bilotta, A.J.; Ma, C.; Huang, X.; Yang, W.; Yao, S.; Cong, Y. Microbiota metabolites SCFA stimulate epithelial migration to promote wound healing through MFGE8 and PAK1. J. Immunol. 2019, 202 (Suppl. 1), 191.15. [Google Scholar]
- Zhao, Y.; Chen, F.; Wu, W.; Sun, M.; Bilotta, A.J.; Yao, S.; Xiao, Y.; Huang, X.; Eaves-Pyles, T.D.; Golovko, G.; et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol. 2018, 11, 752. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Wang, P.; Huang, Y.; Wang, F. Short-Chain Fatty Acids Manifest Stimulative and Protective Effects on Intestinal Barrier Function Through the Inhibition of NLRP3 Inflammasome and Autophagy. Cell. Physiol. Biochem. 2018, 49, 190–205. [Google Scholar] [CrossRef]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor-Dependent Repression of Claudin-2. J. Immunol. 2017, 199, 2976–2984. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Kotani, T.; Konno, T.; Setiawan, J.; Kitamura, Y.; Imada, S.; Usui, Y.; Hatano, N.; Shinohara, M.; Saito, Y.; et al. Promotion of Intestinal Epithelial Cell Turnover by Commensal Bacteria: Role of Short-Chain Fatty Acids. PLoS ONE 2016, 11, e0156334. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-Chain Fatty Acids Activate GPR41 and GPR43 on Intestinal Epithelial Cells to Promote Inflammatory Responses in Mice. Gastroenterology 2013, 145, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Heerdt, B.G.; Houston, M.A.; Augenlicht, L.H. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994, 54, 3288–3293. [Google Scholar] [PubMed]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–g65. [Google Scholar] [CrossRef]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-Modulated Metabolites at the Interface of Host Immunity. J. Immunol. 2017, 198, 572. [Google Scholar] [CrossRef]
- Nastasi, C.; Candela, M.; Bonefeld, C.M.; Geisler, C.; Hansen, M.; Krejsgaard, T.; Biagi, E.; Andersen, M.H.; Brigidi, P.; Ødum, N.; et al. The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci. Rep. 2015, 5, 16148. [Google Scholar] [CrossRef]
- Gonçalves, P.; Araújo, J.R.; Di Santo, J.P. A Cross-Talk Between Microbiota-Derived Short-Chain Fatty Acids and the Host Mucosal Immune System Regulates Intestinal Homeostasis and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 558–572. [Google Scholar] [CrossRef] [Green Version]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef]
- Bhatt, B.; Zeng, P.; Zhu, H.; Sivaprakasam, S.; Xiao, H.; Dong, L.; Shiao, P.; Kolhe, R.; Li, H.; Ganapathy, V.; et al. Gpr109a limits microbiota-induced IL-23 production to constrain ILC3-mediated colonic inflammation and carcinogenesis. J. Immunol. 2018, 200 (Suppl. 1), 2905–2914. [Google Scholar] [CrossRef]
- Docampo, M.D.; Stein-Thoeringer, C.; Lazrak, A.; da Silva, M.B.; van den Brink, M.R.M. Expression of the Butyrate/niacin Receptor, GPR109a on T cells Plays an Important Role in a Mouse Model of Graft Versus Host Disease. J. Immunol. 2019, 202 (Suppl. 1), 69.34. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Segain, J.P.; Raingeard de la Blétière, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.M.; Galmiche, J.P. Butyrate inhibits inflammatory responses through NFkappaB inhibition: Implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Kinoshita, M.; Suzuki, Y.; Saito, Y. Butyrate reduces colonic paracellular permeability by enhancing PPARgamma activation. Biochem. Biophys. Res. Commun. 2002, 293, 827–831. [Google Scholar] [CrossRef]
- Kim, S.V.; Xiang, W.V.; Kwak, C.; Yang, Y.; Lin, X.W.; Ota, M.; Sarpel, U.; Rifkin, D.B.; Xu, R.; Littman, D.R. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 2013, 340, 1456–1459. [Google Scholar] [CrossRef]
- Moreira, J.M.A.; Scheipers, P.; Sørensen, P. The histone deacetylase inhibitor Trichostatin A modulates CD4+ T cell responses. BMC Cancer 2003, 3, 30. [Google Scholar] [CrossRef]
- Kurita-Ochiai, T.; Hashizume, T.; Yonezawa, H.; Ochiai, K.; Yamamoto, M. Characterization of the effects of butyric acid on cell proliferation, cell cycle distribution and apoptosis. FEMS Immunol. Med. Microbiol. 2006, 47, 67–74. [Google Scholar] [CrossRef]
- Zhang, H.; Du, M.; Yang, Q.; Zhu, M.J. Butyrate suppresses murine mast cell proliferation and cytokine production through inhibiting histone deacetylase. J. Nutr. Biochem. 2016, 27, 299–306. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Lee, J.-M.; Lee, E.-J.; Hwang, W.-B.; Kim, D.-J. Indole-3-Carbinol Promotes Goblet-Cell Differentiation Regulating Wnt and Notch Signaling Pathways AhR-Dependently. Mol. Cells 2018, 41, 290–300. [Google Scholar]
- Kimura, A.; Naka, T.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 2009, 206, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Masuda, K.; Kimura, A.; Hanieh, H.; Nguyen, N.T.; Nakahama, T.; Chinen, I.; Otoyo, Y.; Murotani, T.; Yamatodani, A.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages. Int. Immunol. 2011, 23, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Sekine, H.; Mimura, J.; Oshima, M.; Okawa, H.; Kanno, J.; Igarashi, K.; Gonzalez, F.J.; Ikuta, T.; Kawajiri, K.; Fujii-Kuriyama, Y. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol. Cell. Biol. 2009, 29, 6391–6400. [Google Scholar] [CrossRef]
- Kimura, A.; Abe, H.; Tsuruta, S.; Chiba, S.; Fujii-Kuriyama, Y.; Sekiya, T.; Morita, R.; Yoshimura, A. Aryl hydrocarbon receptor protects against bacterial infection by promoting macrophage survival and reactive oxygen species production. Int. Immunol. 2014, 26, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural Aryl Hydrocarbon Receptor Ligands Control Organogenesis of Intestinal Lymphoid Follicles. Science 2011, 334, 1561. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Naka, T.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9721–9726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katepalli, M.; Mueller, C.; Steinmeyer, S.; Jayaraman, A.; Alaniz, R. The microbiota-derived signal indole regulates T-cell fate by reciprocal control of Th17 and Treg development (P3178). J. Immunol. 2013, 190 (Suppl. 1), 61.14. [Google Scholar]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous Stimuli Maintain Intraepithelial Lymphocytes via Aryl Hydrocarbon Receptor Activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Dossa, A.Y.; Escobar, O.; Golden, J.; Frey, M.R.; Ford, H.R.; Gayer, C.P. Bile acids regulate intestinal cell proliferation by modulating EGFR and FXR signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G81–G92. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef]
- Parker, H.E.; Wallis, K.; le Roux, C.W.; Wong, K.Y.; Reimann, F.; Gribble, F.M. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br. J. Pharmacol. 2012, 165, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wammers, M.; Schupp, A.-K.; Bode, J.G.; Ehlting, C.; Wolf, S.; Deenen, R.; Köhrer, K.; Häussinger, D.; Graf, D. Reprogramming of pro-inflammatory human macrophages to an anti-inflammatory phenotype by bile acids. Sci. Rep. 2018, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.W.H.; Puchner, T.; Korkmaz, H.I.; Vos, M.; Soeters, M.R.; de Vries, C.J.M. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS ONE 2017, 12, e0176715. [Google Scholar] [CrossRef] [PubMed]
- Ginty, D.D.; Osborne, D.L.; Seidel, E.R. Putrescine stimulates DNA synthesis in intestinal epithelial cells. Am. J. Physiol. 1989, 257 Pt 1, G145–G150. [Google Scholar] [CrossRef]
- Ray, R.M.; McCormack, S.A.; Covington, C.; Viar, M.J.; Zheng, Y.; Johnson, L.R. The requirement for polyamines for intestinal epithelial cell migration is mediated through Rac1. J. Biol. Chem. 2003, 278, 13039–13046. [Google Scholar] [CrossRef]
- Rao, J.N.; Liu, S.V.; Zou, T.; Liu, L.; Xiao, L.; Zhang, X.; Bellavance, E.; Yuan, J.X.J.; Wang, J.-Y. Rac1 promotes intestinal epithelial restitution by increasing Ca2+ influx through interaction with phospholipase C-γ1 after wounding. Am. J. Physiol. -Cell Physiol. 2008, 295, C1499–C1509. [Google Scholar] [CrossRef]
- Greco, S.; Niepceron, E.; Hugueny, I.; George, P.; Louisot, P.; Biol, M.C. Dietary spermidine and spermine participate in the maturation of galactosyltransferase activity and glycoprotein galactosylation in rat small intestine. J. Nutr. 2001, 131, 1890–1897. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Borovikova, L.V.; Wang, H.; Metz, C.; Tracey, K.J. Spermine inhibition of monocyte activation and inflammation. Mol. Med. 1999, 5, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Caragine, T.; Wang, H.; Cohen, P.S.; Botchkina, G.; Soda, K.; Bianchi, M.; Ulrich, P.; Cerami, A.; Sherry, B.; et al. Spermine inhibits proinflammatory cytokine synthesis in human mononuclear cells: A counterregulatory mechanism that restrains the immune response. J. Exp. Med. 1997, 185, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cano, F.J.; González-Castro, A.; Castellote, C.; Franch, À.; Castell, M. Influence of breast milk polyamines on suckling rat immune system maturation. Dev. Comp. Immunol. 2010, 34, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Jiang, S.; Jiang, Z.; Zheng, C.; Gou, Z. Effects of equol on H2O2-induced oxidative stress in primary chicken intestinal epithelial cells. Poult. Sci. 2016, 95, 1380–1386. [Google Scholar] [CrossRef]
- Di Cagno, R.; Mazzacane, F.; Rizzello, C.G.; Vincentini, O.; Silano, M.; Giuliani, G.; De Angelis, M.; Gobbetti, M. Synthesis of Isoflavone Aglycones and Equol in Soy Milks Fermented by Food-Related Lactic Acid Bacteria and Their Effect on Human Intestinal Caco-2 Cells. J. Agric. Food Chem. 2010, 58, 10338–10346. [Google Scholar] [CrossRef]
- Harada, K.; Sada, S.; Sakaguchi, H.; Takizawa, M.; Ishida, R.; Tsuboi, T. Bacterial metabolite S-equol modulates glucagon-like peptide-1 secretion from enteroendocrine L cell line GLUTag cells via actin polymerization. Biochem. Biophys. Res. Commun. 2018, 501, 1009–1015. [Google Scholar] [CrossRef]
- Wang, C.-W.; Huang, Y.-C.; Chan, F.-N.; Su, S.-C.; Kuo, Y.-H.; Huang, S.-F.; Hung, M.-W.; Lin, H.-C.; Chang, W.-L.; Chang, T.-C. A gut microbial metabolite of ginsenosides, compound K, induces intestinal glucose absorption and Na+/glucose cotransporter 1 gene expression through activation of cAMP response element binding protein. Mol. Nutr. Food Res. 2015, 59, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wan, J.-Y.; Zeng, J.; Huang, W.-H.; Sava-Segal, C.; Li, L.; Niu, X.; Wang, Q.; Wang, C.-Z.; Yuan, C.-S. Effects of compound K, an enteric microbiome metabolite of ginseng, in the treatment of inflammation associated colon cancer. Oncol. Lett. 2018, 15, 8339–8348. [Google Scholar] [CrossRef]
- Lee, I.K.; Kang, K.A.; Lim, C.M.; Kim, K.C.; Kim, H.S.; Kim, D.H.; Kim, B.J.; Chang, W.Y.; Choi, J.H.; Hyun, J.W. Compound K, a metabolite of ginseng saponin, induces mitochondria-dependent and caspase-dependent apoptosis via the generation of reactive oxygen species in human colon cancer cells. Int. J. Mol. Sci. 2010, 11, 4916–4931. [Google Scholar] [CrossRef]
- Mace, T.A.; Ware, M.B.; King, S.A.; Loftus, S.; Farren, M.R.; McMichael, E.; Scoville, S.; Geraghty, C.; Young, G.; Carson, W.E.; et al. Soy isoflavones and their metabolites modulate cytokine-induced natural killer cell function. Sci. Rep. 2019, 9, 5068. [Google Scholar] [CrossRef] [Green Version]
- Gou, Z.; Jiang, S.; Zheng, C.; Tian, Z.; Lin, X. Equol Inhibits LPS-Induced Oxidative Stress and Enhances the Immune Response in Chicken HD11 Macrophages. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Furoku, S.; Nakamoto, M.; Shuto, E.; Hosaka, T.; Nishioka, Y.; Sone, S. The Soy Isoflavone Equol Enhances Antigen-Specific IgE Production in Ovalbumin-Immunized BALB/c Mice. J. Nutr. Sci. Vitaminol. 2010, 56, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Joh, E.-H.; Lee, I.-A.; Jung, I.-H.; Kim, D.-H. Ginsenoside Rb1 and its metabolite compound K inhibit IRAK-1 activation—The key step of inflammation. Biochem. Pharmacol. 2011, 82, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, M.; Hu, S.; Liu, K.; Tai, Y.; Tao, J.; Zhou, W.; Zhao, Z.; Wang, Q.; Wei, W. Ginsenoside metabolite compound-K regulates macrophage function through inhibition of β-arrestin2. Biomed. Pharmacother. 2019, 115, 108909. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Kim, Y.W.; Kim, S.U.; Chae, S.; Koh, Y.S.; Kim, H.S.; Choo, M.K.; Kim, D.H.; Hyun, J.W. G1 phase arrest of the cell cycle by a ginseng metabolite, compound K, in U937 human monocytic leukamia cells. Arch. Pharm. Res. 2005, 28, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, H.; Wang, Q.; Chang, Y.; Liu, K.; Wei, W. Ginsenoside Metabolite Compound K Suppresses T-Cell Priming via Modulation of Dendritic Cell Trafficking and Costimulatory Signals, Resulting in Alleviation of Collagen-Induced Arthritis. J. Pharmacol. Exp. Ther. 2015, 353, 71. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Byeon, H.; Im, K.; Min, H. Effects of ginsenosides on regulatory T cell differentiation. Food Sci. Biotechnol. 2017, 27, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.M.; Shi, Y.Z.; Cheng, M.; Wang, D.F.; Fan, J.F. Serum IL-6, IL-23 profile and Treg/Th17 peripheral cell populations in pediatric patients with inflammatory bowel disease. Pharmazie 2017, 72, 283–287. [Google Scholar]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhong, W.; Wang, W.; Hu, S.; Yuan, J.; Zhang, B.; Hu, T.; Song, G. Ginsenoside Metabolite Compound K Promotes Recovery of Dextran Sulfate Sodium-Induced Colitis and Inhibits Inflammatory Responses by Suppressing NF-κB Activation. PLoS ONE 2014, 9, e87810. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Kohli, R. Bile acid metabolism and signaling: Potential therapeutic target for nonalcoholic fatty liver disease. Clin. Transl. Gastroenterol. 2018, 9, 164. [Google Scholar] [CrossRef]
- Kondo, T.; Kishi, M.; Fushimi, T.; Kaga, T. Acetic acid upregulates the expression of genes for fatty acid oxidation enzymes in liver to suppress body fat accumulation. J. Agric. Food Chem. 2009, 57, 5982–5986. [Google Scholar] [CrossRef]
- Sahuri-Arisoylu, M.; Brody, L.P.; Parkinson, J.R.; Parkes, H.; Navaratnam, N.; Miller, A.D.; Thomas, E.L.; Frost, G.; Bell, J.D. Reprogramming of hepatic fat accumulation and ‘browning’ of adipose tissue by the short-chain fatty acid acetate. Int. J. Obes. 2016, 40, 955–963. [Google Scholar] [CrossRef]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Fukunishi, S.; Sujishi, T.; Takeshita, A.; Ohama, H.; Tsuchimoto, Y.; Asai, A.; Tsuda, Y.; Higuchi, K. Lipopolysaccharides accelerate hepatic steatosis in the development of nonalcoholic fatty liver disease in Zucker rats. J. Clin. Biochem. Nutr. 2014, 54, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hong, S.-W.; Rhee, E.-J.; Lee, W.-Y. GLP-1 Receptor Agonist and Non-Alcoholic Fatty Liver Disease. Diabetes Metab. J. 2012, 36, 262–267. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Rolo, A.P.; Palmeira, C.M. Hepatic FXR: Key regulator of whole-body energy metabolism. Trends Endocrinol. Metab. TEM 2011, 22, 458–466. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.M.; Reimann, F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef]
- Ramos-Molina, B.; Queipo-Ortuño, M.I.; Lambertos, A.; Tinahones, F.J.; Peñafiel, R. Dietary and Gut Microbiota Polyamines in Obesity- and Age-Related Diseases. Front. Nutr. 2019, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Sircana, A.; Framarin, L.; Leone, N.; Berrutti, M.; Castellino, F.; Parente, R.; De Michieli, F.; Paschetta, E.; Musso, G. Altered Gut Microbiota in Type 2 Diabetes: Just a Coincidence? Curr. Diabetes Rep. 2018, 18, 98. [Google Scholar] [CrossRef]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediat. Inflamm. 2014, 2014, 162021. [Google Scholar] [CrossRef]
- Mandaliya, D.K.; Seshadri, S. Short Chain Fatty Acids, pancreatic dysfunction and type 2 diabetes. Pancreatology 2019, 19, 280–284. [Google Scholar] [CrossRef]
- Yan, X.; Li, P.; Tang, Z.; Feng, B. The relationship between bile acid concentration, glucagon-like-peptide 1, fibroblast growth factor 15 and bile acid receptors in rats during progression of glucose intolerance. BMC Endocr. Disord. 2017, 17, 60. [Google Scholar] [CrossRef]
- Tuomainen, M.; Lindström, J.; Lehtonen, M.; Auriola, S.; Pihlajamäki, J.; Peltonen, M.; Tuomilehto, J.; Uusitupa, M.; de Mello, V.D.; Hanhineva, K. Associations of serum indolepropionic acid, a gut microbiota metabolite, with type 2 diabetes and low-grade inflammation in high-risk individuals. Nutr. Diabetes 2018, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.M.; Maddox, T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I.; Pankow, J.S.; Ballantyne, C.M.; Couper, D.; Vigo, A.; Hoogeveen, R.; Folsom, A.R.; Heiss, G. Low-grade systemic inflammation and the development of type 2 diabetes: The atherosclerosis risk in communities study. Diabetes 2003, 52, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Wahbi, K. CNS-disease affecting the heart: Brain–heart disorders. J. Neurol. Sci. 2014, 345, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Soda, K.; Kano, Y.; Chiba, F. Food polyamine and cardiovascular disease—An epidemiological study. Glob. J. Health Sci. 2012, 4, 170–178. [Google Scholar] [CrossRef]
- Morgan, D.M. Uptake of polyamines by human endothelial cells. Characterization and lack of effect of agonists of endothelial function. Biochem. J. 1992, 286 Pt 2, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Khurana, S.; Raufman, J.P.; Pallone, T.L. Bile acids regulate cardiovascular function. Clin. Transl. Sci. 2011, 4, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H. Cardiovascular Diseases and Panax ginseng: A Review on Molecular Mechanisms and Medical Applications. J. Ginseng Res. 2012, 36, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobionda, S.; Sittipo, P.; Kwon, H.Y.; Lee, Y.K. The Role of Gut Microbiota in Intestinal Inflammation with Respect to Diet and Extrinsic Stressors. Microorganisms 2019, 7, 271. [Google Scholar] [CrossRef]
- Vamanu, E.; Pelinescu, D.; Gatea, F.; Sarbu, I. Altered in Vitro Metabolomic Response of the Human Microbiota to Sweeteners. Genes 2019, 10, 535. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Microbial Metabolites | Effect on IECs |
|---|---|
| SCFAs | Promote epithelial barrier function; Induce IEC proliferation and turnover mediated by activation of MEK–ERK through GPR41 or GPR43; Enhance IEC differentiation and apoptosis; Activate cytokine production via MEK–ERK and p38 MAPK signaling mediated by GPR41 or GPR43; Promote the production of antimicrobial peptides via the activation of mTOR and STAT3 signaling mediated by GPR43; Increase wound healing by stimulating IEC migration through PAKI and MFGE8; Increase the secretion of GLP-1 by L cells. |
| AHR ligands | Increase tight junction gene expression; Influence IEC differentiation by suppressing enterocyte differentiation and inducing secretory cell differentiation; Decrease pro-inflammatory cytokine production and increase anti-inflammatory cytokine production by attenuating NF-κB. |
| Bile acids | Regulate IEC integrity by interacting with FXR; Alter colonic cell proliferation and apoptosis depending on concentration; Enhance GLP-1 secretion by L-cells by activating TGR5 and cAMP signaling. |
| Polyamines | Enhance DNA synthesis; Induce cell migration through Rac1 activation and calcium influx; Promote IEC maturation by increasing glycoprotein galactosylation; Suppress the production of IL-18 and anti-microbial peptides by reducing NLRP6 inflammasome assembly. |
| Equol | Protect IECs from oxidative damage by promoting the expression of antioxidant genes, enhancing antioxidant enzyme activity; Maintain the integrity of the tight junctions and inhibit IL-8 production; Increase intracellular Ca2+ levels, actin reorganization, suppression of GLP-1 secretion by enteroendocrine L cells through GPR30. |
| Compound K | Enhance SGLT1-mediated glucose uptake by inducing CREB and CBP binding to the SGLT1 promoter; Inhibit IL-8 secretion by LPS-activated IECs; Inhibit cell growth by arresting the cell cycle in G1 phase; Induce caspase-dependent apoptosis via ROS generation. |
| Microbial Metabolites | Effect on Immune Cells |
|---|---|
| SCFAs | Inhibit NF-κB-mediated pro-inflammatory cytokine expression; Modulate recruitment of immune cells to the intestinal tract by controlling the expression of chemokines or chemoattractant receptors; Inhibit immune cell proliferation and induce GPR41-dependent cell apoptosis; Reduce the expression of co-stimulatory molecules and induce anti-inflammatory properties in antigen-presenting cells; Contribute to Treg differentiation through increased histone H3 acetylation of Foxp3 promoter region in a GPR43-dependent manner; Promote homing of Tregs in the colon in a GPR15-dependent manner. |
| AHR ligands | Inhibit production of pro-inflammatory cytokines such as IL-1β and IL-6 by macrophages; Inhibit migration of macrophages toward chemokines; Induce the expansion and IL-22 production by ILC3; Promote bacterial clearance by macrophages via inducing survival and ROS production; Regulate Treg/Th17 lineage fate by inducing expansion of Tregs, while suppressing Th17 development; Maintain IELs in the intestinal tract. |
| Bile acids | Block caspase-1 maturation and IL-1 and IL-18 secretion from LPS-primed BMDMs via the TGR5–cAMP–PKA axis; Reprogram pro-inflammatory macrophages to an anti-inflammatory macrophage; Inhibit Th1 activation by binding VDR and inhibiting ERK1/2 phosphorylation. |
| Polyamines | Inhibit pro-inflammatory cytokine synthesis in LPS-activated monocytes and macrophages; Accelerate intestinal and systemic immune cell maturation, e.g. T cells and B cells. |
| Equol | Decrease the production of IL-12/IL-18-induced IFN-γ production by natural killer cells; Protect macrophages from LPS-induced oxidative stress by reducing lipid peroxidation and enhancing activity of antioxidant enzymes; Induce antigen-specific IgE production from B cells and IL-13 production from T cells. |
| Compound K | Inhibit pro-inflammatory cytokine production in LPS-activated macrophages by inhibiting NF-κB; Inhibit macrophage function including polarization and phagocytosis by altering β-arrestin 2 coupling; Induce cell cycle arrest in the G1 phase and apoptosis; Suppress T cell priming by inhibiting the trafficking and signals for T cell activation by dendritic cells. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sittipo, P.; Shim, J.-w.; Lee, Y.K. Microbial Metabolites Determine Host Health and the Status of Some Diseases. Int. J. Mol. Sci. 2019, 20, 5296. https://doi.org/10.3390/ijms20215296
Sittipo P, Shim J-w, Lee YK. Microbial Metabolites Determine Host Health and the Status of Some Diseases. International Journal of Molecular Sciences. 2019; 20(21):5296. https://doi.org/10.3390/ijms20215296
Chicago/Turabian StyleSittipo, Panida, Jae-won Shim, and Yun Kyung Lee. 2019. "Microbial Metabolites Determine Host Health and the Status of Some Diseases" International Journal of Molecular Sciences 20, no. 21: 5296. https://doi.org/10.3390/ijms20215296
APA StyleSittipo, P., Shim, J. -w., & Lee, Y. K. (2019). Microbial Metabolites Determine Host Health and the Status of Some Diseases. International Journal of Molecular Sciences, 20(21), 5296. https://doi.org/10.3390/ijms20215296