rAAV Engineering for Capsid-Protein Enzyme Insertions and Mosaicism Reveals Resilience to Mutational, Structural and Thermal Perturbations

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results
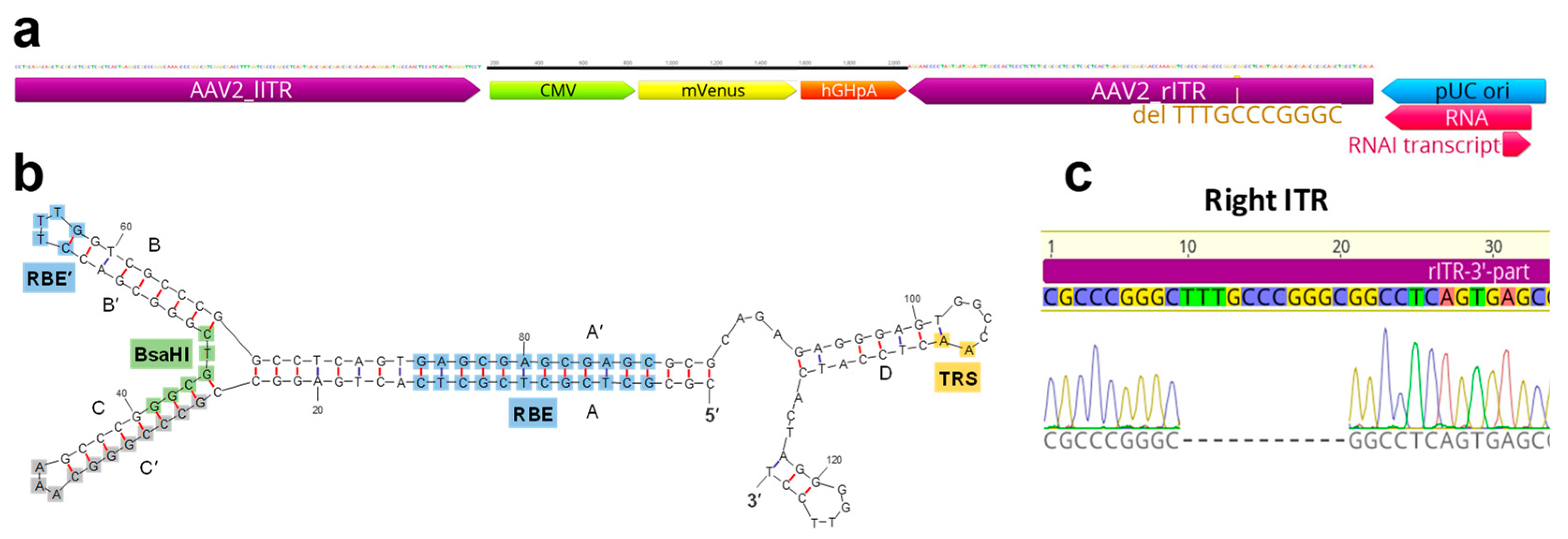
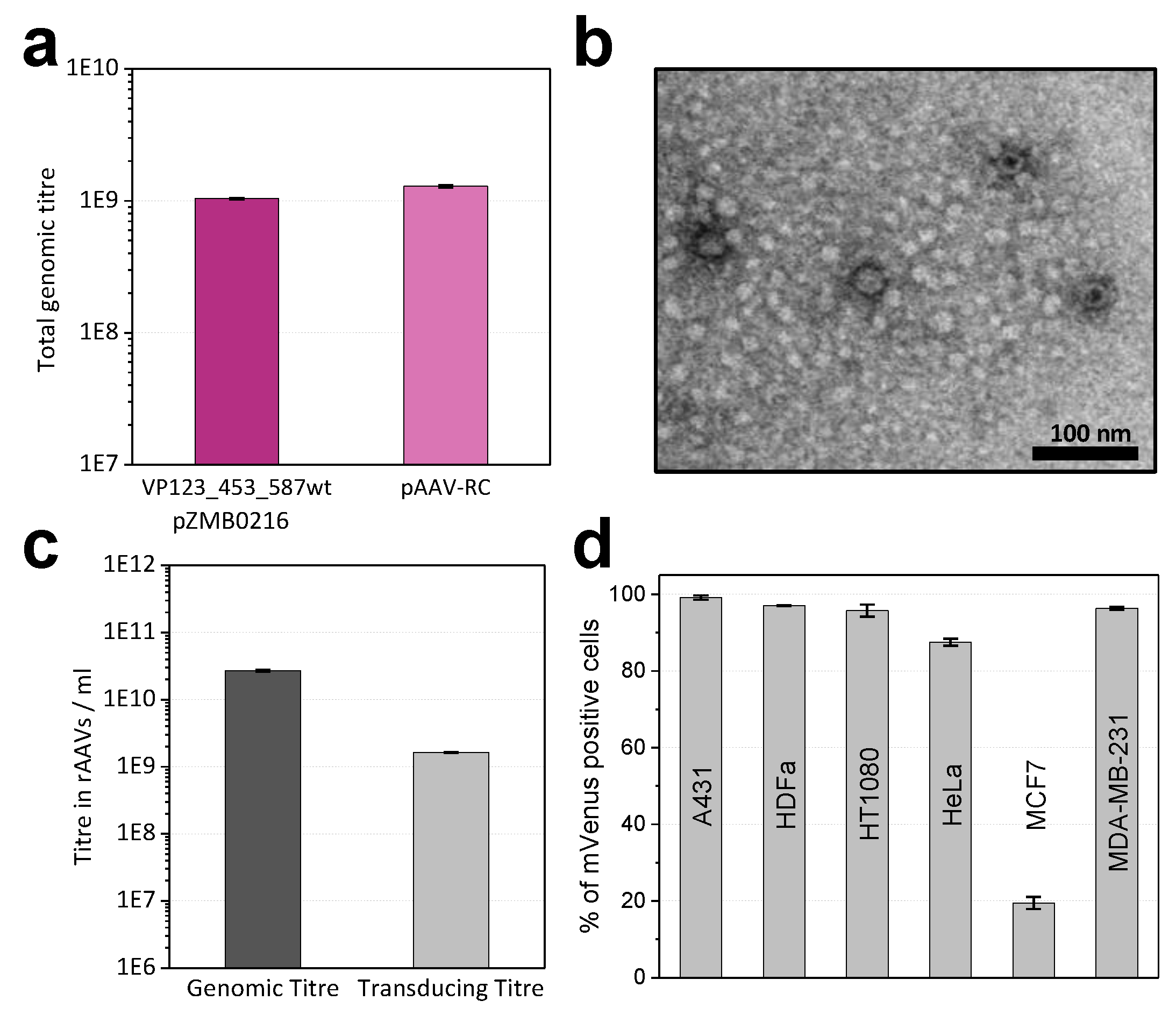
2.1. Modifications of ITR and RepCap Plasmids Are Compatible with rAAV Production
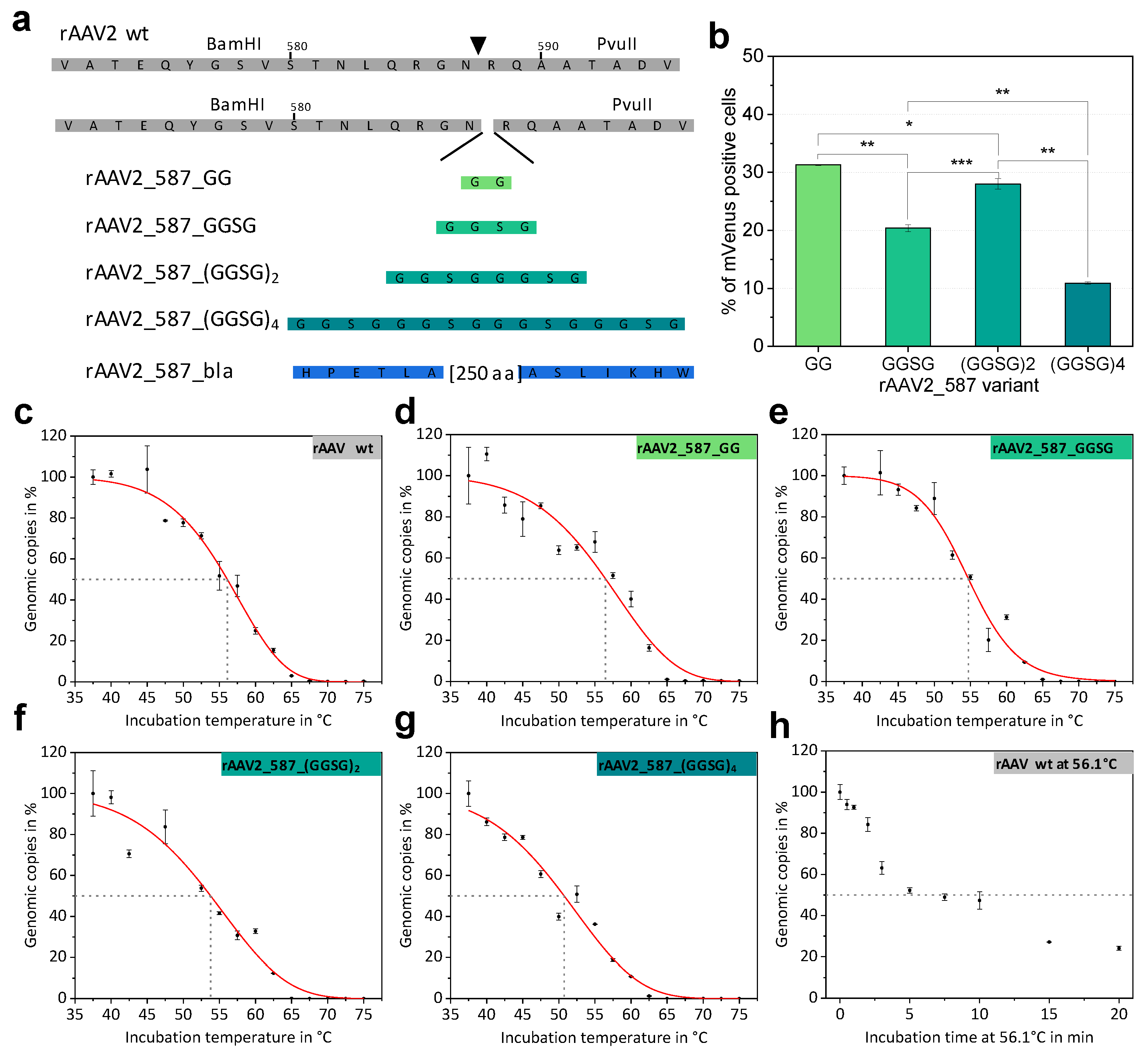
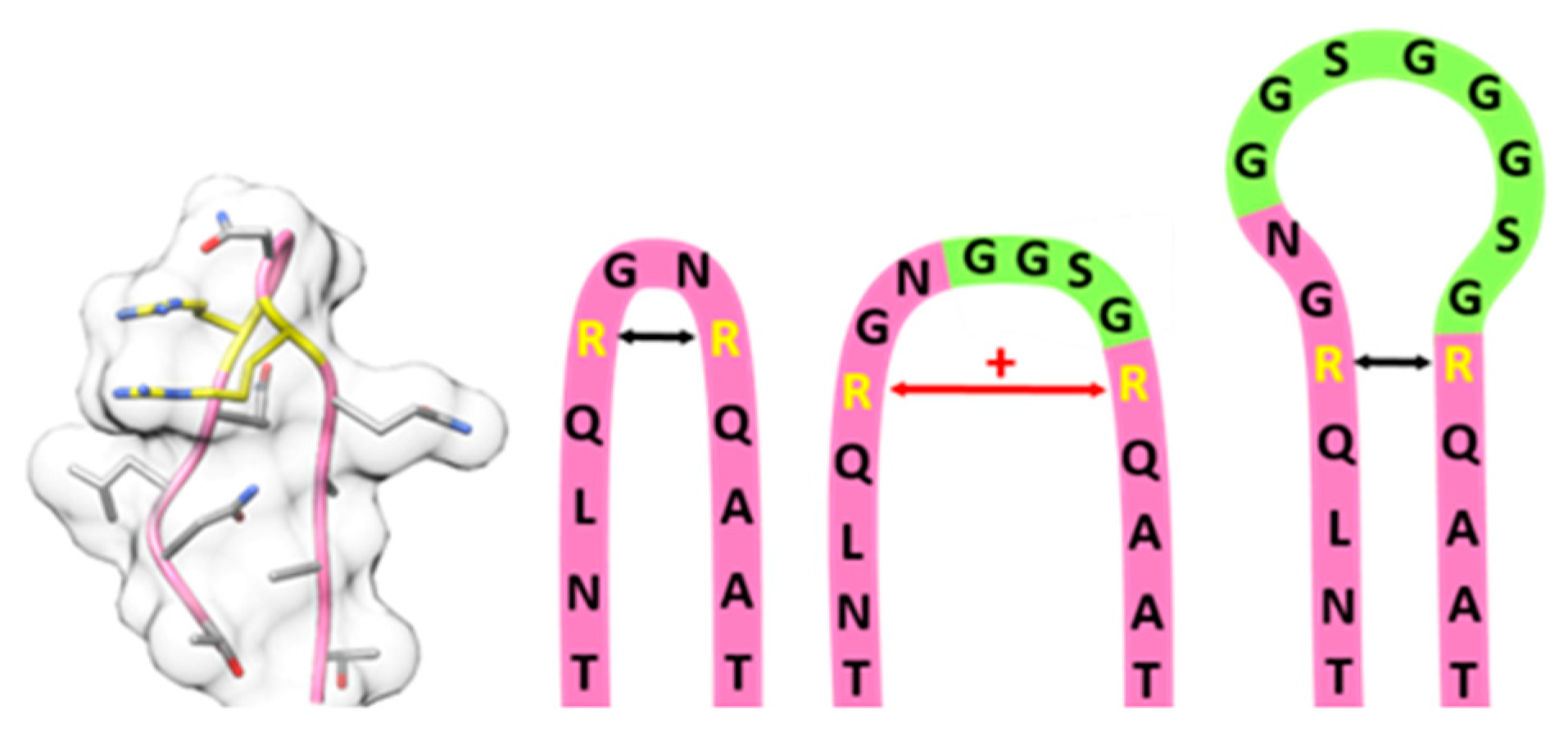
2.2. Systematic Variation of Loop Modifications Shows A Complex Pattern of Stability and Transduction Efficiency
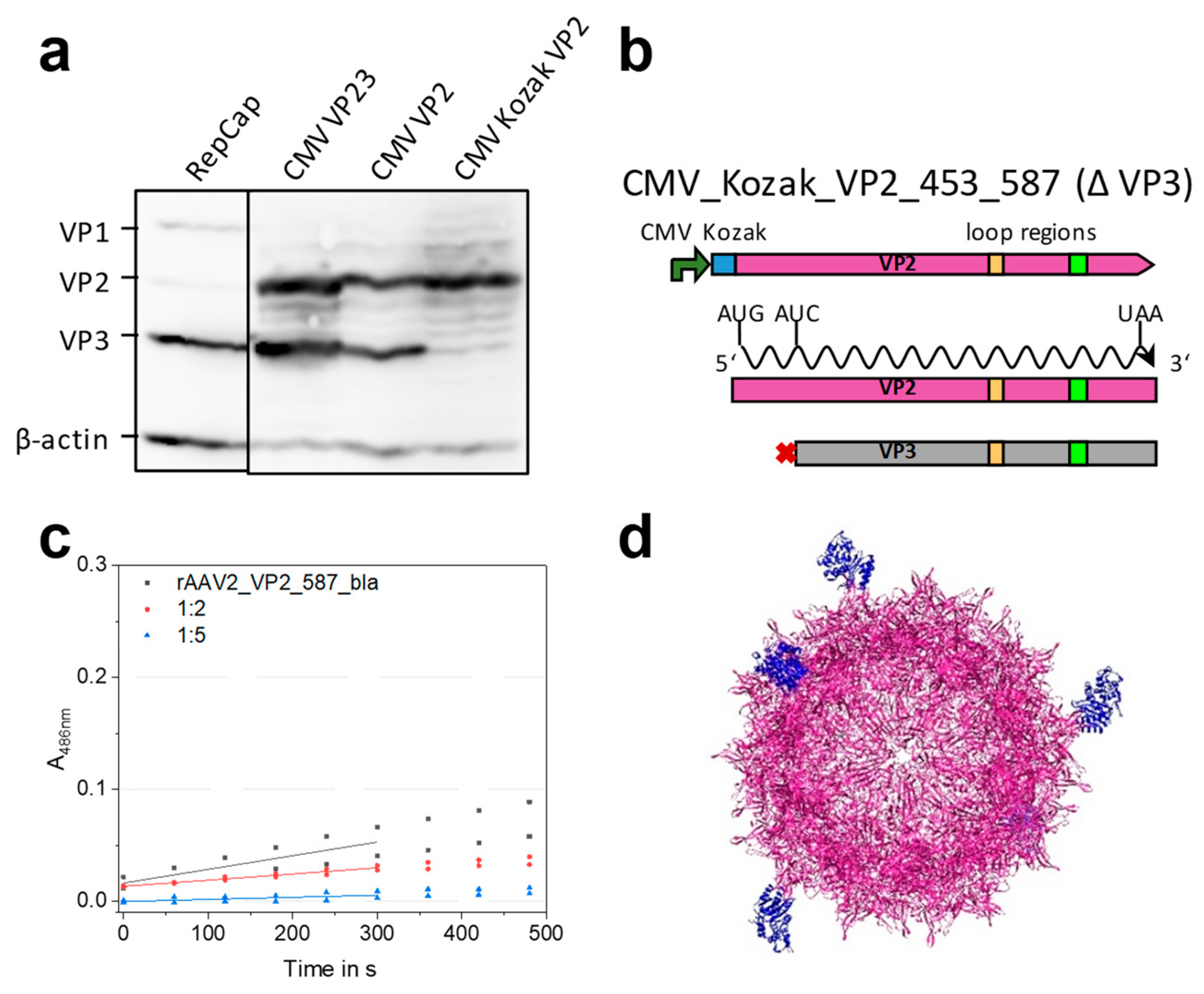
2.3. Mosaic rAAVs with a 29 kDa β-Lactamase at Position 578 in VP2 Require a Kozak Consensus Sequence
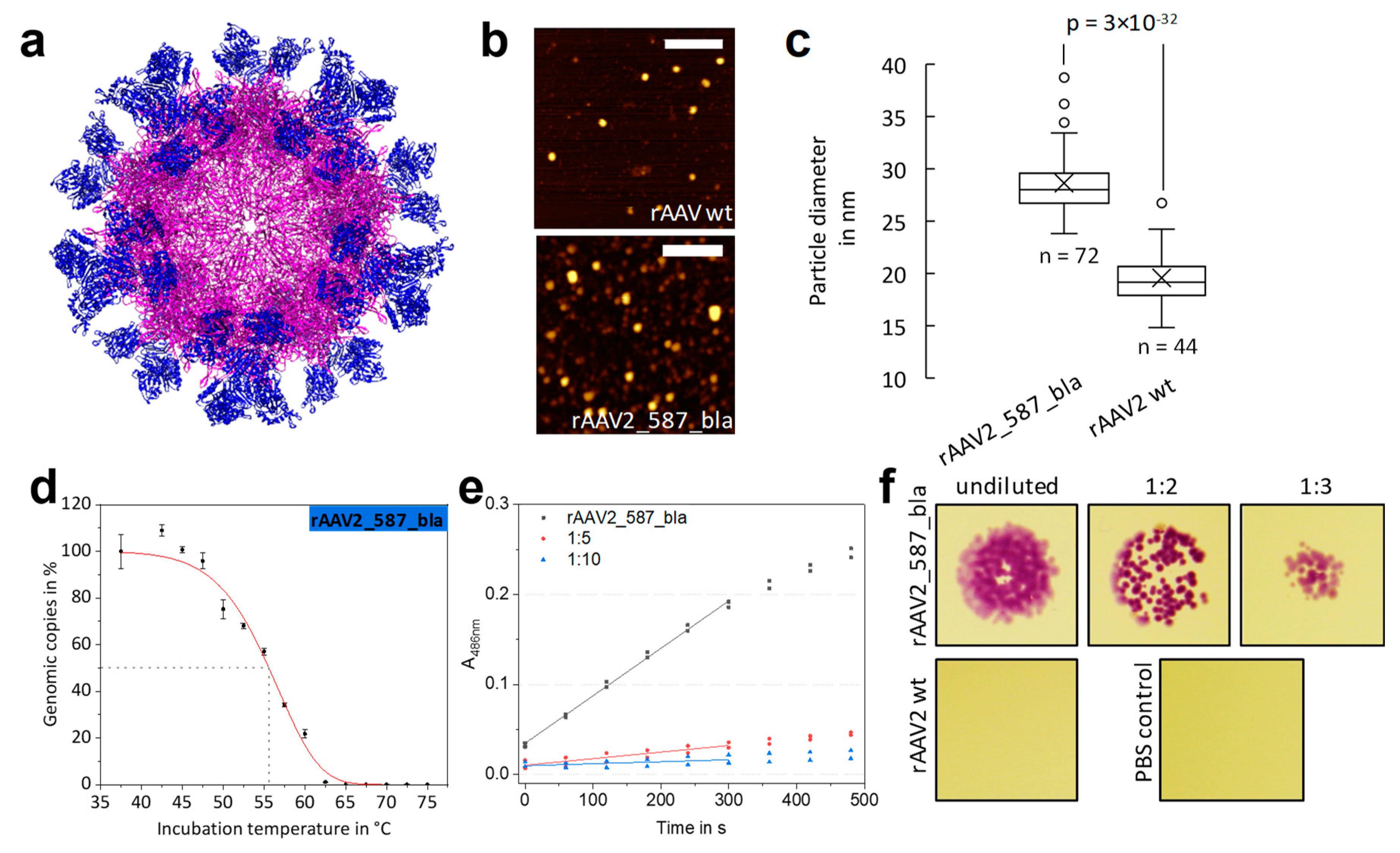
2.4. Fully Lactamase-Decorated rAAV Capsids Can Be Produced and Show Enzyme Activity
3. Discussion
4. Materials and Methods
4.1. Construction of Plasmids
4.2. Cell Culture
4.3. Viral Particle Production
4.4. Purification of Viral Particles
4.5. SDS-PAGE and Western Blot Analysis
4.6. Determination of Genomic Titers
4.7. Transducing Titer Assays
4.8. Transmission Electron Microscopy
4.9. Atomic Force Microscopy
4.10. AAV Stability Assay
4.11. β-Lactamase Activity Assays
4.12. Statistical Analysis and Reproducibility
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| rAAV | Recombinant Adeno-associated virus |
| TEM | Transmission electron microscopy |
| AFM | Atomic force microscopy |
| bla | β-lactamase |
References
- FDA Advisory Committee. Briefing Document; Luxturna™ (voretigene neparvovec); Spark Therapeutics, Inc.: Hampton, VA, USA, 2017. [Google Scholar]
- Clément, N.; Grieger, J.C. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16002. [Google Scholar] [CrossRef]
- Xie, Q.; Bu, W.; Bhatia, S.; Hare, J.; Somasundaram, T.; Azzi, A.; Chapman, M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 10405–10410. [Google Scholar] [CrossRef]
- Rose, J.A.; Maizel, J.V.; Inman, J.K.; Shatkin, A.J. Structural proteins of adenovirus-associated viruses. J. Virol. 1971, 8, 766–770. [Google Scholar]
- Trempe, J.P.; Carter, B.J. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J. Virol. 1988, 62, 3356–3363. [Google Scholar]
- Stutika, C.; Gogol-Döring, A.; Botschen, L.; Mietzsch, M.; Weger, S.; Feldkamp, M.; Chen, W.; Heilbronn, R. A Comprehensive RNA Sequencing Analysis of the Adeno-Associated Virus (AAV) Type 2 Transcriptome Reveals Novel AAV Transcripts, Splice Variants, and Derived Proteins. J. Virol. 2016, 90, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Elliger, S.; Elliger, C.; Podsakoff, G.; Villarreal, L.; Kurtzman, G.J.; Iwaki, Y.; Colosi, P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998, 5, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, J.; Samulski, R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar] [PubMed]
- Grimm, D.; Kay, M.A.; Kleinschmidt, J.A. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 2003, 7, 839–850. [Google Scholar] [CrossRef]
- Feiner, R.C.; Müller, K.M. Recent progress in protein-protein interaction study for EGFR-targeted therapeutics. Expert Rev. Proteomics 2016, 13, 817–832. [Google Scholar] [CrossRef]
- Grimm, D.; Kay, M. a From virus evolution to vector revolution: Use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr. Gene Ther. 2003, 3, 281–304. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Lux, K.; Goerlitz, N.; Schlemminger, S.; Perabo, L.; Goldnau, D.; Endell, J.; Leike, K.; Kofler, D.M.; Finke, S.; Hallek, M. Green Fluorescent Protein-Tagged Adeno-Associated Virus Particles Allow the Study of Cytosolic and Nuclear Trafficking. J. Virol. 2005, 79, 11776–11787. [Google Scholar] [CrossRef] [PubMed]
- Münch, R.C.; Janicki, H.; Völker, I.; Rasbach, A.; Hallek, M.; Büning, H.; Buchholz, C.J. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 2013, 21, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hagen, S.; Baumann, T.; Wagner, H.J.; Morath, V.; Kaufmann, B.; Fischer, A.; Bergmann, S.; Schindler, P.; Arndt, K.M.; Müller, K.M. Modular adeno-associated virus (rAAV) vectors used for cellular virus-directed enzyme prodrug therapy. Sci. Rep. 2014, 4, 3759. [Google Scholar] [CrossRef]
- Girod, A.; Ried, M.; Wobus, C.; Lahm, H.; Leike, K.; Kleinschmidt, J.; Deléage, G.; Hallek, M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 1999, 5, 1438. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Bartlett, J.S. RGD inclusion in VP3 provides adeno-associated virus type 2 (AAV2)-based vectors with a heparan sulfate-independent cell entry mechanism. Mol. Ther. 2003, 7, 515–525. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, Y.; Zhou, Y.; Zandi, E.; Lee, C.-L.; Joo, K.-I.; Wang, P. Site-specific modification of adeno-associated viruses via a genetically engineered aldehyde tag. Small 2013, 9, 421–429. [Google Scholar] [CrossRef]
- Falck, G.; Müller, K.M. Enzyme-Based Labeling Strategies for Antibody–Drug Conjugates and Antibody Mimetics. Antibodies 2018, 7, 4. [Google Scholar] [CrossRef]
- Ried, M.U.; Girod, A.; Leike, K.; Büning, H.; Hallek, M. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J. Virol. 2002, 76, 4559–4566. [Google Scholar] [CrossRef]
- Nieto, K.; Weghofer, M.; Sehr, P.; Ritter, M.; Sedlmeier, S.; Karanam, B.; Seitz, H.; Müller, M.; Kellner, M.; Hörer, M.; et al. Development of AAVLP(HPV16/31L2) particles as broadly protective HPV vaccine candidate. PLoS ONE 2012, 7, e39741. [Google Scholar] [CrossRef]
- Judd, J.; Wei, F.; Nguyen, P.Q.; Tartaglia, L.J.; Agbandje-McKenna, M.; Silberg, J.J.; Suh, J. Random Insertion of mCherry Into VP3 Domain of Adeno-associated Virus Yields Fluorescent Capsids With no Loss of Infectivity. Mol. Ther. Nucleic Acids 2012, 1, e54. [Google Scholar] [CrossRef] [PubMed]
- Freiburg iGEM Team Virus Construction Kit For Therapy. Available online: http://2010.igem.org/Team:Freiburg_Bioware (accessed on 24 January 2019).
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; El-Gohary, Y.; Prasadan, K.; Shiota, C.; Xiao, X.; Wiersch, J.; Paredes, J.; Tulachan, S.; Gittes, G.K. Rapid and simplified purification of recombinant adeno-associated virus. J. Virol. Methods 2012, 183, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Zeltner, N.; Kohlbrenner, E.; Clément, N.; Weber, T.; Linden, R.M. Near-perfect infectivity of wild-type AAV as benchmark for infectivity of recombinant AAV vectors. Gene Ther. 2010, 17, 872–879. [Google Scholar] [CrossRef]
- Ellis, B.L.; Hirsch, M.L.; Barker, J.C.; Connelly, J.P.; Steininger, R.J.; Porteus, M.H. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol. J. 2013, 10, 74. [Google Scholar] [CrossRef]
- Rayaprolu, V.; Kruse, S.; Kant, R.; Venkatakrishnan, B.; Movahed, N.; Brooke, D.; Lins, B.; Bennett, A.; Potter, T.; McKenna, R.; et al. Comparative Analysis of Adeno Associated Virus Capsid Stability and Dynamics. J. Virol. 2013, 87, 13150–13160. [Google Scholar] [CrossRef]
- Speck, J.; Hecky, J.; Tam, H.K.; Arndt, K.M.; Einsle, O.; Müller, K.M. Exploring the molecular linkage of protein stability traits for enzyme optimization by iterative truncation and evolution. Biochemistry 2012, 51, 4850–4867. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Hecky, J.; Müller, K.M. Structural perturbation and compensation by directed evolution at physiological temperature leads to thermostabilization of β-lactamase. Biochemistry 2005, 44, 12640–12654. [Google Scholar] [CrossRef]
- Samulski, R.J.; Srivastava, A.; Berns, K.I.; Muzyczka, N. Rescue of adeno-associated virus from recombinant plasmids: Gene correction within the terminal repeats of AAV. Cell 1983, 33, 135–143. [Google Scholar] [CrossRef]
- Troll, C.; Yoder, J.; Alexander, D.; Hernández, J.; Loh, Y.; Camps, M. The mutagenic footprint of low-fidelity Pol I ColE1 plasmid replication in E. coli reveals an extensive interplay between Pol I and Pol III. Curr. Genet. 2014, 60, 123–134. [Google Scholar] [CrossRef] [PubMed]
- AAV-MCS expression vector VPK-410; Cell Biolabs, Inc.: San Diego, CA, USA, 2016; Available online: https://www.cellbiolabs.com/sites/default/files/VPK-410%20Sequence_0.doc (accessed on 5 September 2018).
- Xie, J.; Mao, Q.; Tai, P.W.L.; He, R.; Ai, J.; Su, Q.; Zhu, Y.; Ma, H.; Li, J.; Gong, S.; et al. Short DNA Hairpins Compromise Recombinant Adeno-Associated Virus Genome Homogeneity. Mol. Ther. 2017, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Warrington, K.H.; Gorbatyuk, O.S.; Harrison, J.K.; Opie, S.R.; Zolotukhin, S.; Muzyczka, N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 2004, 78, 6595–6609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Arnold, G.S.; Bartlett, J.S. Insertional mutagenesis of the adeno-associated virus type 2 (AAV2) capsid gene and generation of AAV2 vectors targeted to alternative cell-surface receptors. Hum. Gene Ther. 2001, 12, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Schmidt, K.; Leder, C.; Müller, O.J.; Wobus, C.E.; Bettinger, K.; Von der Lieth, C.W.; King, J.A.; Kleinschmidt, J.A. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 2003, 77, 11072–11081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opie, S.R.; Warrington, K.H., Jr.; Agbandje-McKenna, M.; Zolotukhin, S.; Muzyczka, N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 2003, 77, 6995–7006. [Google Scholar] [CrossRef] [Green Version]
- Perabo, L.; Goldnau, D.; White, K.; Endell, J.; Boucas, J.; Humme, S.; Work, L.M.; Janicki, H.; Hallek, M.; Baker, A.H.; et al. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J. Virol. 2006, 80, 7265–7269. [Google Scholar] [CrossRef] [Green Version]
- Ros, C.; Baltzer, C.; Mani, B.; Kempf, C. Parvovirus uncoating in vitro reveals a mechanism of DNA release without capsid disassembly and striking differences in encapsidated DNA stability. Virology 2006, 345, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Bernaud, J.; Rossi, A.; Fis, A.; Gardette, L.; Aillot, L.; Büning, H.; Castelnovo, M.; Salvetti, A.; Faivre-Moskalenko, C. Characterization of AAV vector particle stability at the single-capsid level. J. Biol. Phys. 2018, 44, 181–194. [Google Scholar] [CrossRef]
- Bennett, A.; Patel, S.; Mietzsch, M.; Jose, A.; Lins-Austin, B.; Yu, J.C.; Bothner, B.; McKenna, R.; Agbandje-McKenna, M. Thermal Stability as a Determinant of AAV Serotype Identity. Mol. Ther.-Methods Clin. Dev. 2017, 6, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Xie, J.; Dmitriev, I.; Kashentseva, E.; Curiel, D.T.; Hsu, H.; Mountz, J.D. Addition of six-His-tagged peptide to the C terminus of adeno-associated virus VP3 does not affect viral tropism or production. J. Virol. 2002, 76, 12023–12031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, M. Initiation of translation in prokaryotes and eukaryotes. Gene 1999, 234, 187–208. [Google Scholar] [CrossRef]
- Knight, T. Idempotent Vector Design for Standard Assembly of Biobricks Standard Biobrick Sequence Interface. Available online: http://hdl.handle.net/1721.1/45138 (accessed on 24 January 2019).
- Müller, K.M.; Arndt, K.M.; IGEM_Freiburg; Grünberg, R. Fusion Protein (Freiburg) Biobrick assembly standard. Available online: http://hdl.handle.net/1721.1/45140 (accessed on 24 January 2019).
- Agilent Technologies AAV Helper-Free System Instruction Manual. Available online: https://www.agilent.com/cs/library/usermanuals/Public/240071.pdf (accessed on 13 August 2019).
- Zolotukhin, S.; Byrne, B.J.; Mason, E.; Zolotukhin, I.; Potter, M.; Chesnut, K.; Summerford, C.; Samulski, R.J.; Muzyczka, N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6, 973–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Name with Description | Length | Backbone |
|---|---|---|
| pZMB0522_ITR_EXS_CMV_mVenus_hGHpA AAV2 ITR flanking a CMV promoter expressing the fluorescent protein mVenus | 4014 bp | pUC19 |
| pZMB0216_Rep_VP123_453_587wt_p5tataless expression of VP1/2/3 of AAV2 with cloning ready 453 and 587 loop regions, arginines in 587 loop region are intact, p5 promoter at end of expression cassette | 5455 bp | pSB1C3_001 |
| pZMB0600_Rep_VP13_453_587wt_p5tataless expression of VP1 and VP3 with cloning ready 453 and 587 loop regions Arg in 587 loop region are intact | 6455 bp | pSB1C3_001 |
| pZMB0315_CMV_Kozak_VP2_453_587wtHis expression of VP2 with Kozak sequence to prevent leaky scanning and VP3 start knock out, cloning ready 453 and 587 loop regions, Arg in 587 intact, His-tag in 587 loop | 4705 bp | pSB1C3_001 |
| Sample | Titer in vg/mL a | Transduction Ability in % b | Td, 5 min in °Cc |
|---|---|---|---|
| rAAV2 wt | 3.1 × 1010 | 96.7 ± 0.1 | 56.1 ± 0.5 |
| rAAV2_587_GG | 7.1 × 109 | 31.3 ± 0.1 | 56.4 ± 0.8 |
| rAAV2_587_GGSG | 4.0 × 1010 | 20.4 ± 0.6 | 54.7 ± 0.5 |
| rAAV2_587_(GGSG)2 | 2.4 × 1010 | 28.0 ± 0.9 | 53.8 ± 0.8 |
| rAAV2_587_(GGSG)4 | 4.7 × 109 | 10.9 ± 0.2 | 50.7 ± 0.7 |
| rAAV2_587_bla | 1.3 × 1010 | 1.2 ± 0.1 | 55.6 ± 0.4 |
| rAAV2_VP2_587_bla | 6.3 × 1010 | 57.0 ± 2 | n.d. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feiner, R.C.; Teschner, J.; Teschner, K.E.; Radukic, M.T.; Baumann, T.; Hagen, S.; Hannappel, Y.; Biere, N.; Anselmetti, D.; Arndt, K.M.; et al. rAAV Engineering for Capsid-Protein Enzyme Insertions and Mosaicism Reveals Resilience to Mutational, Structural and Thermal Perturbations. Int. J. Mol. Sci. 2019, 20, 5702. https://doi.org/10.3390/ijms20225702
Feiner RC, Teschner J, Teschner KE, Radukic MT, Baumann T, Hagen S, Hannappel Y, Biere N, Anselmetti D, Arndt KM, et al. rAAV Engineering for Capsid-Protein Enzyme Insertions and Mosaicism Reveals Resilience to Mutational, Structural and Thermal Perturbations. International Journal of Molecular Sciences. 2019; 20(22):5702. https://doi.org/10.3390/ijms20225702
Chicago/Turabian StyleFeiner, Rebecca C., Julian Teschner, Kathrin E. Teschner, Marco T. Radukic, Tobias Baumann, Sven Hagen, Yvonne Hannappel, Niklas Biere, Dario Anselmetti, Katja M. Arndt, and et al. 2019. "rAAV Engineering for Capsid-Protein Enzyme Insertions and Mosaicism Reveals Resilience to Mutational, Structural and Thermal Perturbations" International Journal of Molecular Sciences 20, no. 22: 5702. https://doi.org/10.3390/ijms20225702
APA StyleFeiner, R. C., Teschner, J., Teschner, K. E., Radukic, M. T., Baumann, T., Hagen, S., Hannappel, Y., Biere, N., Anselmetti, D., Arndt, K. M., & Müller, K. M. (2019). rAAV Engineering for Capsid-Protein Enzyme Insertions and Mosaicism Reveals Resilience to Mutational, Structural and Thermal Perturbations. International Journal of Molecular Sciences, 20(22), 5702. https://doi.org/10.3390/ijms20225702