The Integrin Activating Protein Kindlin-3 Is Cleaved in Human Platelets during ST-Elevation Myocardial Infarction

Abstract
:1. Introduction
2. Results
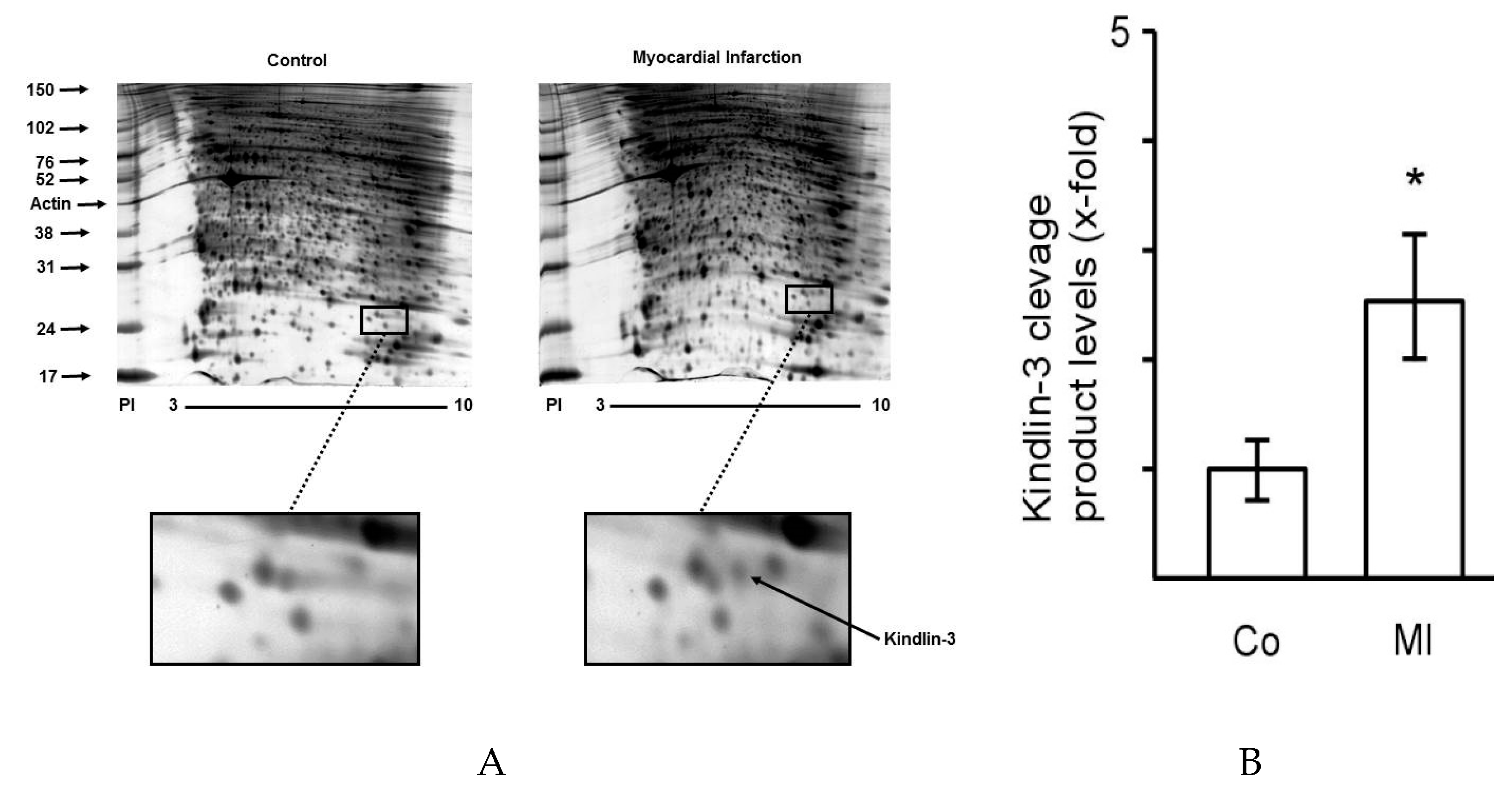
2.1. The Proteome of Platelets from Patients with Myocardial Infarction Shows Distinct Changes
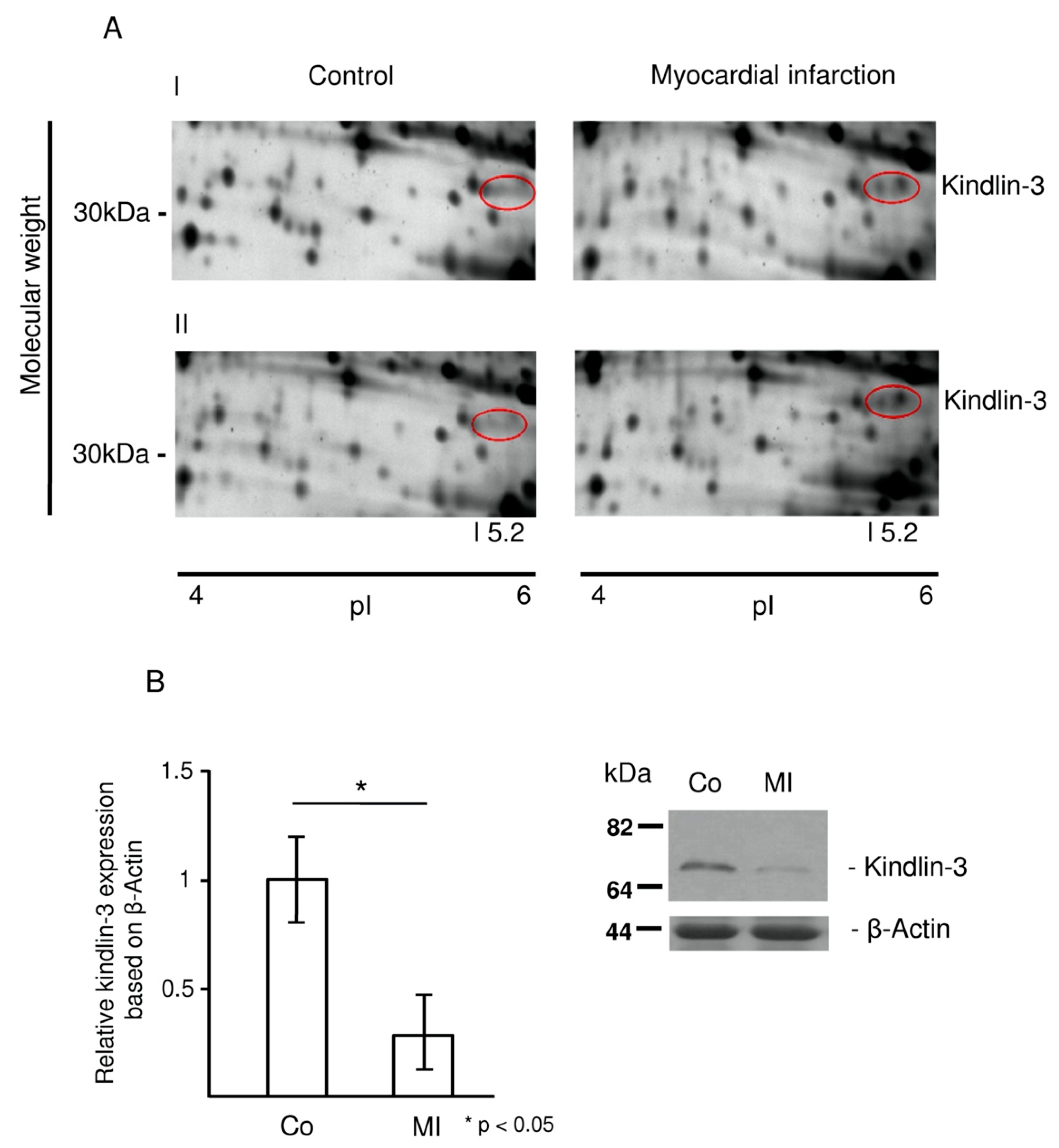
2.2. Kindlin-3 Is Cleaved in Platelets during Myocardial Infarction
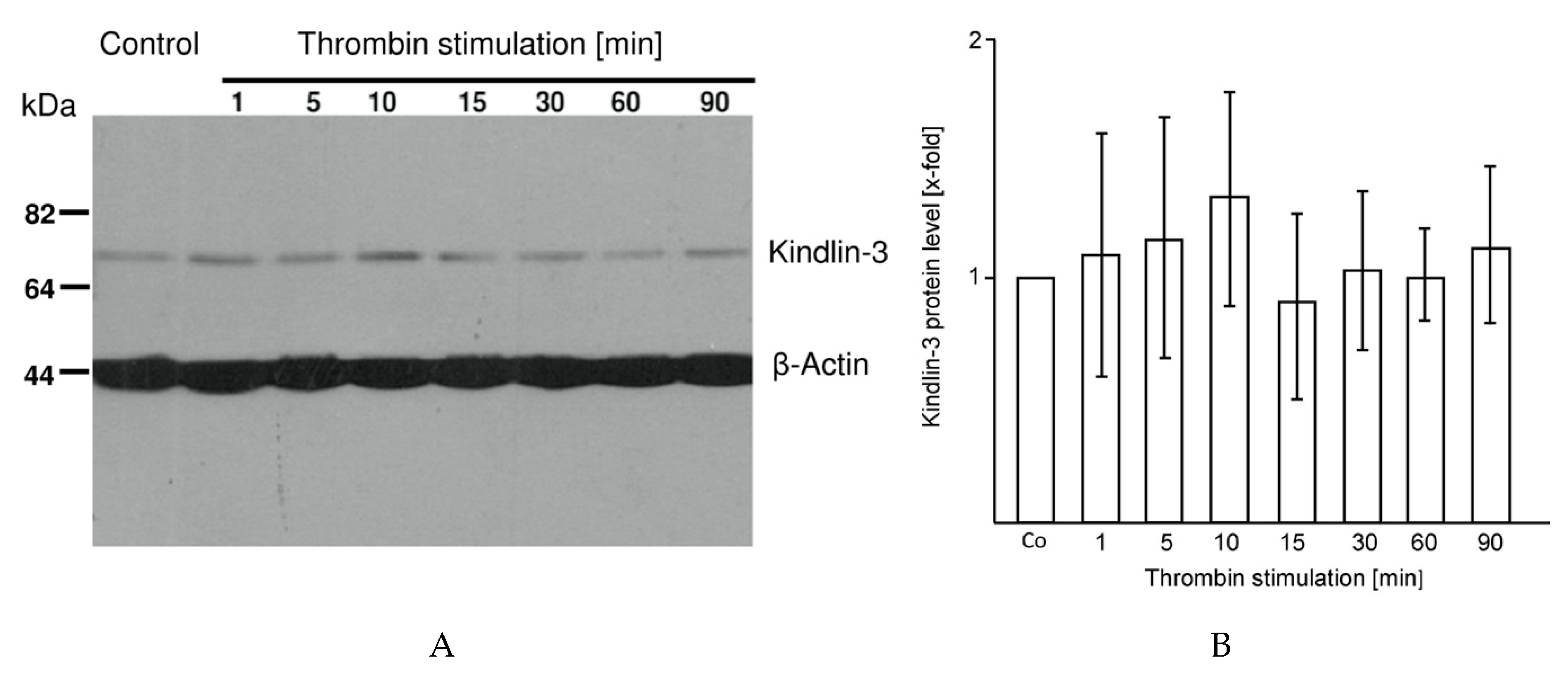
2.3. Kindlin-3 Cleavage Is not Induced by Thrombin Stimulation
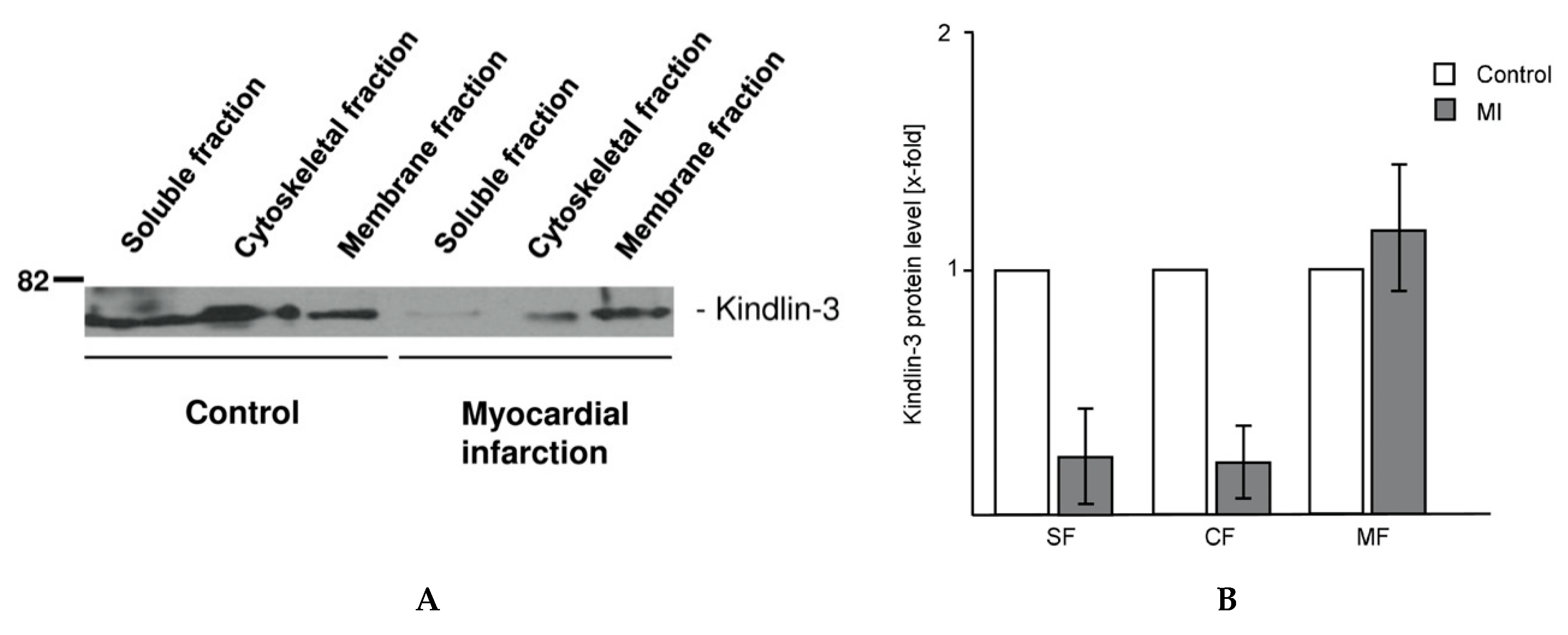
2.4. Kindlin-3 Is Located in the Cytoskeleton, the Plasma Membrane, and the Soluble Fraction of Human Platelets and Becomes Redistributed during Myocardial Infarction
2.5. Patient Characteristics
3. Discussion
4. Material and Methods
4.1. Study Design
4.2. Platelet Isolation
4.3. Fractionation of Soluble, Cytoskeleletal and Platelet Membrane Proteins
4.4. Two-Dimensional Gel Electrophoresis
4.5. Immunoblotting
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ussar, S.; Wang, H.V.; Linder, S.; Fassler, R.; Moser, M. The Kindlins: Subcellular localization and expression during murine development. Exp. Cell Res. 2006, 312, 3142–3151. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; de Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin beta tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Malinin, N.L.; Plow, E.F.; Byzova, T.V. Kindlins in FERM adhesion. Blood 2010, 115, 4011–4017. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Bauer, M.; Schmid, S.; Ruppert, R.; Schmidt, S.; Sixt, M.; Wang, H.V.; Sperandio, M.; Fassler, R. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 2009, 15, 300–305. [Google Scholar] [CrossRef]
- Svensson, L.; Howarth, K.; McDowall, A.; Patzak, I.; Evans, R.; Ussar, S.; Moser, M.; Metin, A.; Fried, M.; Tomlinson, I.; et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 2009, 15, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chen, X.; Zhi, H.; Gao, J.; Bialkowska, K.; Byzova, T.V.; Pluskota, E.; White, G.C.; Liu, J.; Plow, E.F.; et al. Direct interaction of kindlin-3 with integrin alphaIIbbeta3 in platelets is required for supporting arterial thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1961–1967. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Nieswandt, B.; Ussar, S.; Pozgajova, M.; Fassler, R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 2008, 14, 325–330. [Google Scholar] [CrossRef]
- Kruger, M.; Moser, M.; Ussar, S.; Thievessen, I.; Luber, C.A.; Forner, F.; Schmidt, S.; Zanivan, S.; Fassler, R.; Mann, M. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 2008, 134, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Kuijpers, T.W.; van de Vijver, E.; Weterman, M.A.; de Boer, M.; Tool., A.T.; van den Berg, T.K.; Moser, M.; Jakobs, M.E.; Seeger, K.; Sanal, O.; et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood 2009, 113, 4740–4746. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc. Res. 2004, 61, 498–511. [Google Scholar] [CrossRef]
- Gawaz, M.; Neumann, F.J.; Ott, I.; Schiessler, A.; Schomig, A. Platelet function in acute myocardial infarction treated with direct angioplasty. Circulation 1996, 93, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Fox, K.A.; Hacke, W.; Berger, P.B.; Black, H.R.; Boden, W.E.; Cacoub, P.; Cohen., E.A.; Creager, M.A.; Easton, J.D.; et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N. Engl. J. Med. 2006, 354, 1706–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boersma, E.; Harrington, R.A.; Moliterno, D.J.; White, H.; Theroux, P.; Van de Werf, F.; de Torbal, A.; Armstrong, P.W.; Wallentin, L.C.; Wilcox, R.G.; et al. Platelet glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: A meta-analysis of all major randomised clinical trials. Lancet 2002, 359, 189–198. [Google Scholar] [CrossRef]
- Bigalke, B.; Haap, M.; Stellos, K.; Geisler, T.; Seizer, P.; Kremmer, E.; Overkamp, D.; Gawaz, M. Platelet glycoprotein VI (GPVI) for early identification of acute coronary syndrome in patients with chest pain. Thromb. Res. 2010, 125, e184–e189. [Google Scholar] [CrossRef] [PubMed]
- Geisler, T.; Fekecs, L.; Wurster, T.; Chiribiri, A.; Schuster, A.; Nagel, E.; Miller, S.; Gawaz, M.; Stellos, K.; Bigalke, B. Association of platelet-SDF-1 with hemodynamic function and infarct size using cardiac MR in patients with AMI. Eur. J. Radiol. 2012, 81, e486–e490. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.D.; Barnard, M.R.; Krueger, L.A.; Valeri, C.R.; Furman, M.I. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: Studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001, 104, 1533–1537. [Google Scholar] [CrossRef] [Green Version]
- Stellos, K.; Bigalke, B.; Langer, H.; Geisler, T.; Schad, A.; Kogel, A.; Pfaff, F.; Stakos, D.; Seizer, P.; Muller, I.; et al. Expression of stromal-cell-derived factor-1 on circulating platelets is increased in patients with acute coronary syndrome and correlates with the number of CD34+ progenitor cells. Eur. Heart J. 2009, 30, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Malinin, N.L.; Meller, J.; Ma, Y.; West, X.Z.; Bledzka, K.; Qin, J.; Podrez, E.A.; Byzova, T.V. Regulation of cell adhesion and migration by Kindlin-3 cleavage by calpain. J. Biol. Chem. 2012, 287, 40012–40020. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Li, W.; Willard, B.; Silverstein, R.L.; McIntyre, T.M. Proteasome proteolysis supports stimulated platelet function and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Grundler, K.; Rotter, R.; Tilley, S.; Pircher, J.; Czermak, T.; Yakac, M.; Gaitzsch, E.; Massberg, S.; Krotz, F.; Sohn, H.Y.; et al. The proteasome regulates collagen-induced platelet aggregation via nuclear-factor-kappa-B (NFkB) activation. Thromb. Res. 2016, 148, 15–22. [Google Scholar] [CrossRef]
- Oksala, N.; Parssinen, J.; Seppala, I.; Klopp, N.; Illig, T.; Laaksonen, R.; Levula, M.; Raitoharju, E.; Kholova, I.; Sioris, T.; et al. Kindlin 3 (FERMT3) is associated with unstable atherosclerotic plaques, anti-inflammatory type II macrophages and upregulation of beta-2 integrins in all major arterial beds. Atherosclerosis 2015, 242, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Mory, A.; Feigelson, S.W.; Yarali, N.; Kilic, S.S.; Bayhan, G.I.; Gershoni-Baruch, R.; Etzioni, A.; Alon, R. Kindlin-3: A new gene involved in the pathogenesis of LAD-III. Blood 2008, 112, 2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.H.; Ashton, G.H.; Penagos, H.G.; Lee, J.V.; Feiler, H.S.; Wilhelmsen, K.C.; South, A.P.; Smith, F.J.; Prescott, A.R.; Wessagowit, V.; et al. Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am. J. Hum. Genet. 2003, 73, 174–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, H.; Umeshita, K.; Sakon, M.; Imajoh-Ohmi, S.; Fujitani, K.; Gotoh, M.; Oiki, E.; Kambayashi, J.; Monden, M. Calpain activation in plasma membrane bleb formation during tert-butyl hydroperoxide-induced rat hepatocyte injury. Gastroenterology 1996, 110, 1897–1904. [Google Scholar] [CrossRef]
- Pasquet, J.M.; Dachary-Prigent, J.; Nurden, A.T. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur. J. Biochem. 1996, 239, 647–654. [Google Scholar] [CrossRef]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Franks, Z.G.; Vieira de Abreu, A.; Grundler, K.; Kile, B.T.; Dhakal, B.K.; Rondina, M.T.; Kahr, W.H.; et al. Bacteria differentially induce degradation of Bcl-xL, a survival protein, by human platelets. Blood 2012, 120, 5014–5020. [Google Scholar] [CrossRef]
- Herz, C.; Aumailley, M.; Schulte, C.; Schlotzer-Schrehardt, U.; Bruckner-Tuderman, L.; Has, C. Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. J. Biol. Chem. 2006, 281, 36082–36090. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Wu, S.; Shi, X.; Chen, K.; Wu, C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 2003, 113, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Block, M.R.; Badowski, C.; Millon-Fremillon, A.; Bouvard, D.; Bouin, A.P.; Faurobert, E.; Gerber-Scokaert, D.; Planus, E.; Albiges-Rizo, C. Podosome-type adhesions and focal adhesions, so alike yet so different. Eur. J. Cell Biol. 2008, 87, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, B.F.; Borst, O.; Gehring, E.M.; Schoenberger, T.; Urban, B.; Ninci, E.; Seizer, P.; Schmidt, C.; Bigalke, B.; Koch, M.; et al. PI3 kinase-dependent stimulation of platelet migration by stromal cell-derived factor 1 (SDF-1). J. Mol. Med. 2010, 88, 1277–1288. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Kraemer, B.F.; Borst, O.; Munzer, P.; Schonberger, T.; Schmidt, C.; Leibrock, C.; Towhid, S.T.; Seizer, P.; Kuhl, D.; et al. SGK1 sensitivity of platelet migration. Cell Physiol. Biochem. 2012, 30, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.F.; Schmidt, C.; Urban, B.; Bigalke, B.; Schwanitz, L.; Koch, M.; Seizer, P.; Schaller, M.; Gawaz, M.; Lindemann, S. High shear flow induces migration of adherent human platelets. Platelets 2011, 22, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; Elstad, M.R.; McEver, R.P.; McIntyre, T.M.; Moore, K.L.; Morrissey, J.H.; Prescott, S.M.; Zimmerman, G.A. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Investig. 1996, 97, 1525–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffert, Y.; Franz-Wachtel, M.; Fladerer, C.; Nordheim, A.; Reuther, J.; Wohlleben, W.; Mast, Y. Proteomic analysis of the GlnR-mediated response to nitrogen limitation in Streptomyces coelicolor M145. Appl. Microbiol. Biotechnol. 2011, 89, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Hala, M.; Cole, R.; Synek, L.; Drdova, E.; Pecenkova, T.; Nordheim, A.; Lamkemeyer, T.; Madlung, J.; Hochholdinger, F.; Fowler, J.E.; et al. An exocyst complex functions in plant cell growth in Arabidopsis and tobacco. Plant Cell 2008, 20, 1330–1345. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Group (n = 12) | Myocardial Infarction (STEMI) (n = 12) | |
|---|---|---|
| Age (years, mean ± SEM) | 65 ± 3 | 67 ± 4 |
| Arterial Hypertension; n= | 6 | 7 |
| Diabetes mellitus; n= | 3 | 2 |
| Smoking; n= | 6 | 7 |
| Impaired renal function; n= | 2 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraemer, B.F.; Lamkemeyer, T.; Franz-Wachtel, M.; Lindemann, S. The Integrin Activating Protein Kindlin-3 Is Cleaved in Human Platelets during ST-Elevation Myocardial Infarction. Int. J. Mol. Sci. 2019, 20, 6154. https://doi.org/10.3390/ijms20246154
Kraemer BF, Lamkemeyer T, Franz-Wachtel M, Lindemann S. The Integrin Activating Protein Kindlin-3 Is Cleaved in Human Platelets during ST-Elevation Myocardial Infarction. International Journal of Molecular Sciences. 2019; 20(24):6154. https://doi.org/10.3390/ijms20246154
Chicago/Turabian StyleKraemer, Bjoern F., Tobias Lamkemeyer, Mirita Franz-Wachtel, and Stephan Lindemann. 2019. "The Integrin Activating Protein Kindlin-3 Is Cleaved in Human Platelets during ST-Elevation Myocardial Infarction" International Journal of Molecular Sciences 20, no. 24: 6154. https://doi.org/10.3390/ijms20246154
APA StyleKraemer, B. F., Lamkemeyer, T., Franz-Wachtel, M., & Lindemann, S. (2019). The Integrin Activating Protein Kindlin-3 Is Cleaved in Human Platelets during ST-Elevation Myocardial Infarction. International Journal of Molecular Sciences, 20(24), 6154. https://doi.org/10.3390/ijms20246154