Expression of Maize MADS Transcription Factor ZmES22 Negatively Modulates Starch Accumulation in Rice Endosperm


Abstract
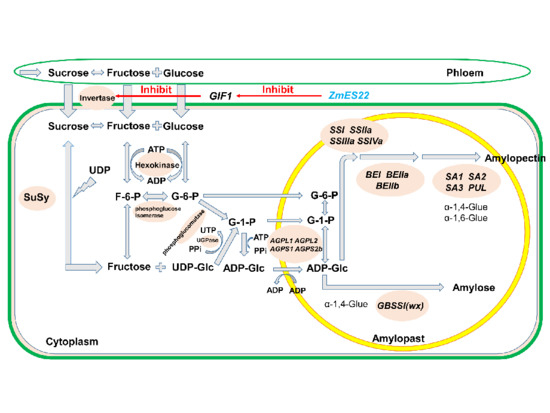
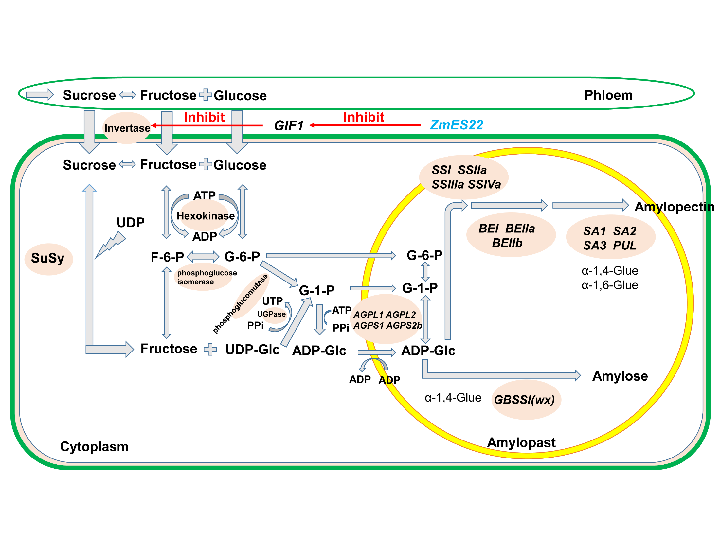
:
1. Introduction
2. Results
2.1. Sequence Analysis and Construction of Phylogenetic Tree for ZmES22 Homologues
2.2. Expression Profiles and Subcellular Localization of ZmES22
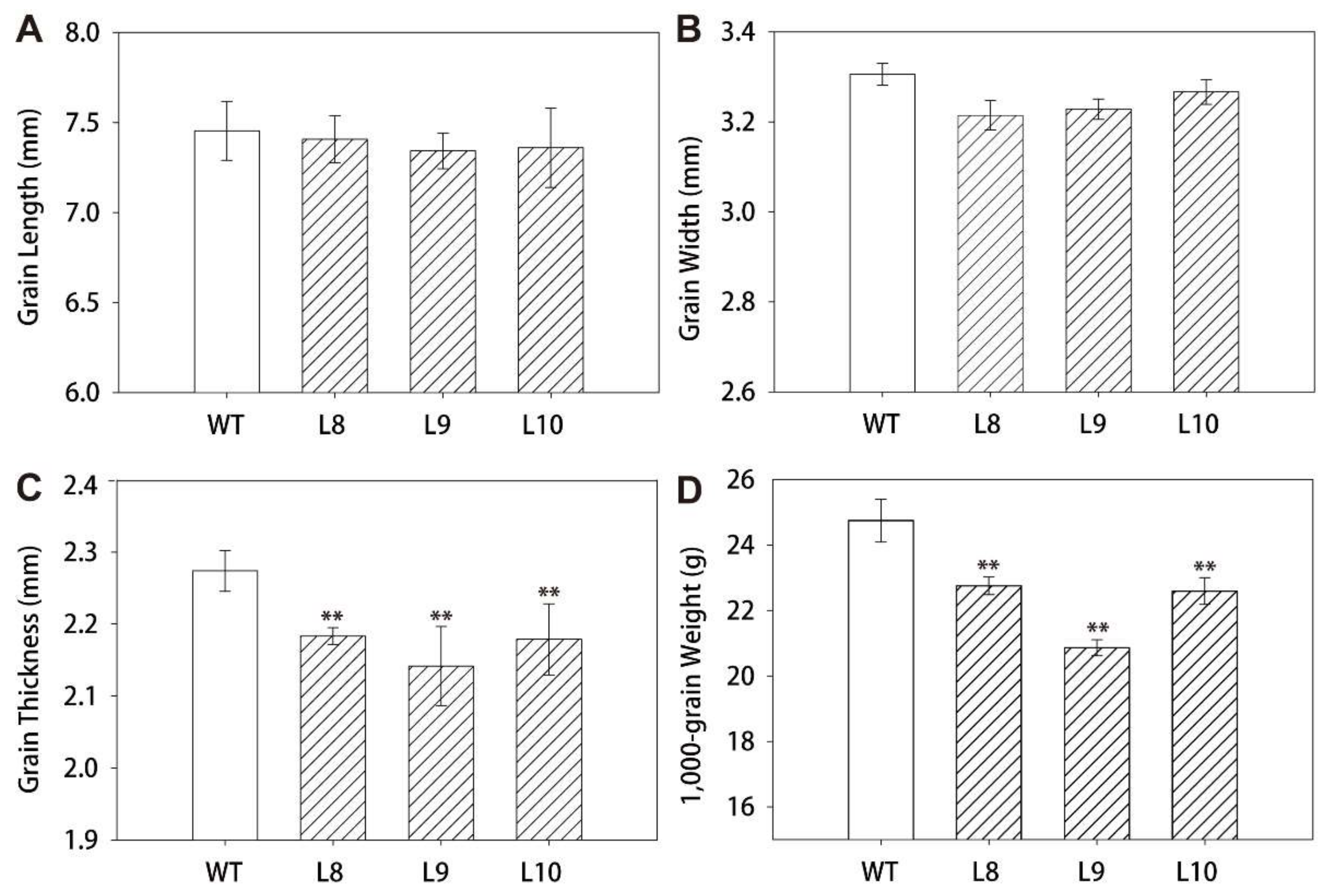
2.3. Analysis of Agronomic Characters of ZmES22 Overexpression Transgenic Rice
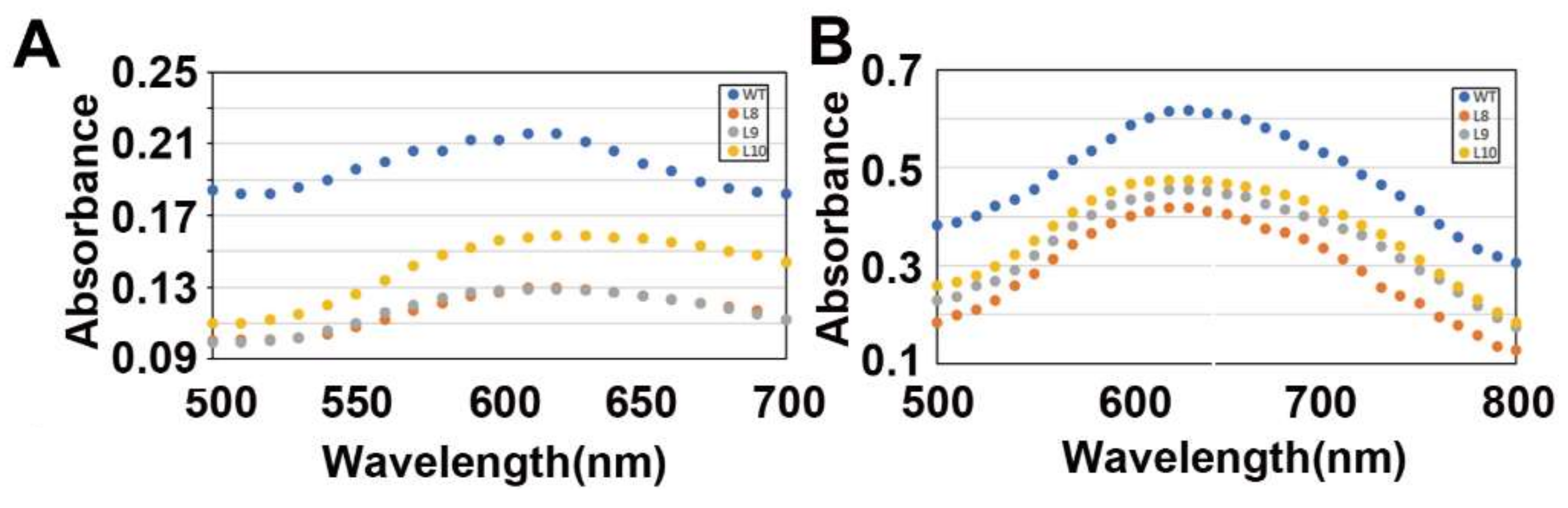
2.4. Overexpression of ZmES22 Influences Starch Structure in Transgenic Rice
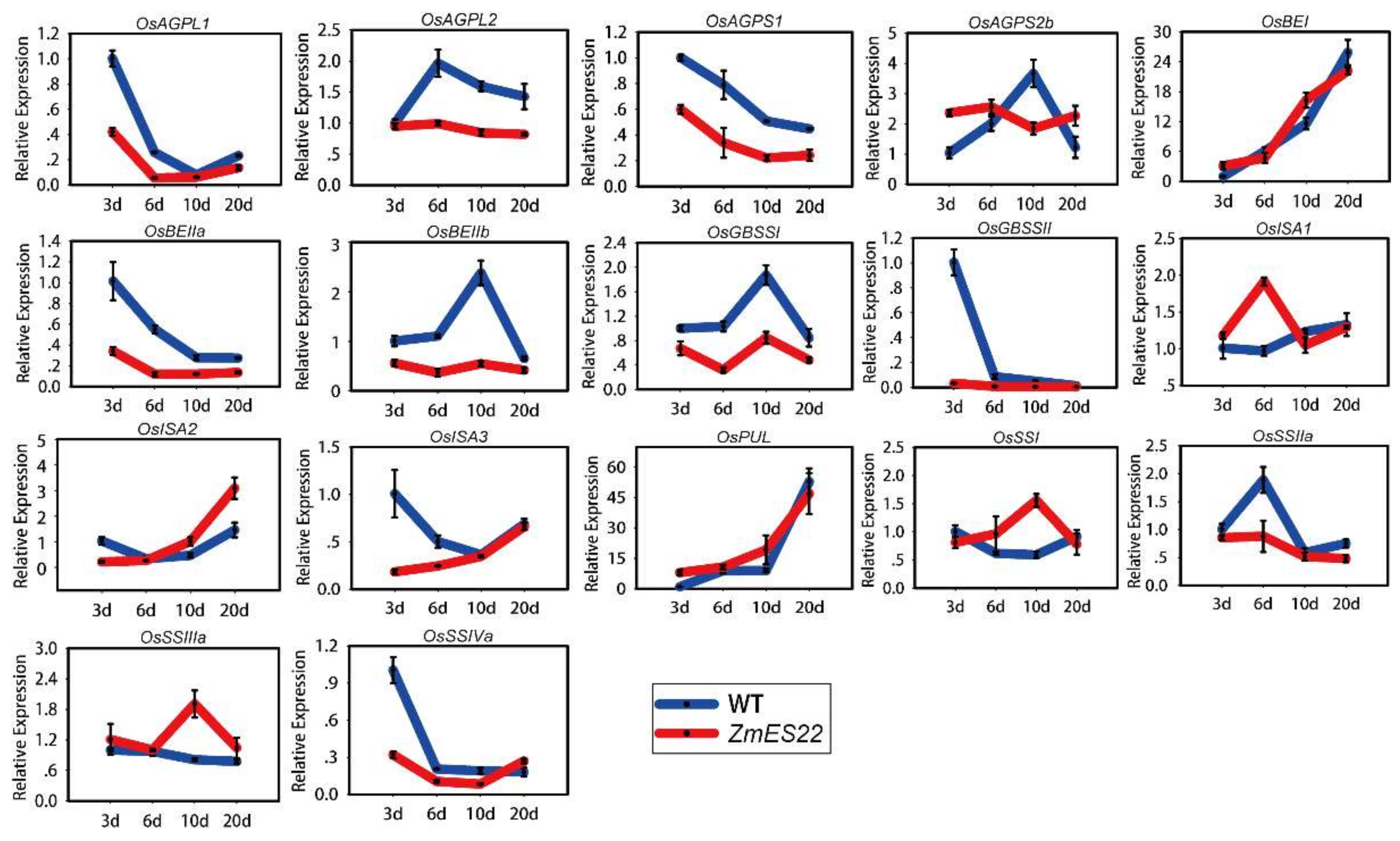
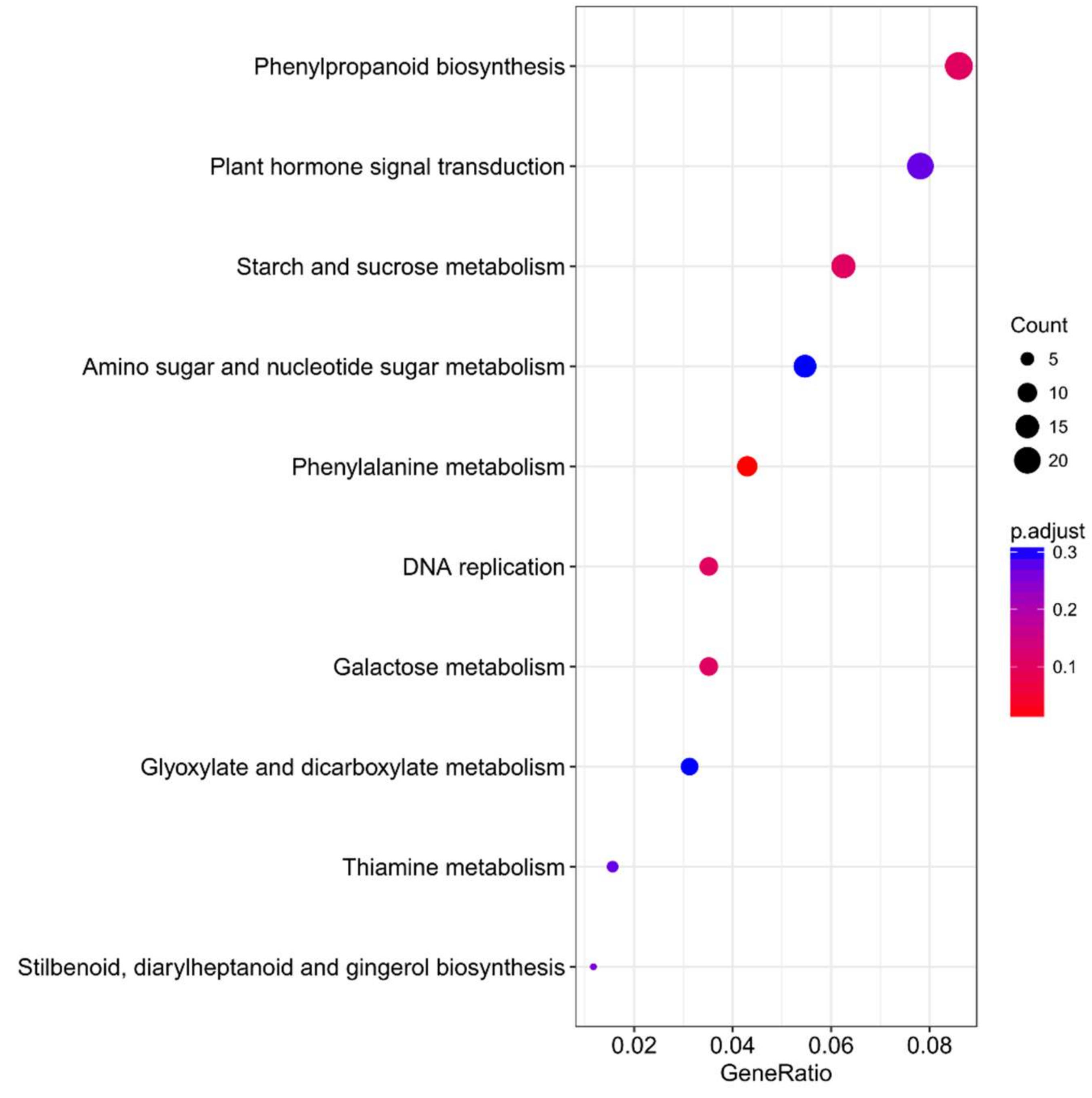
2.5. Overexpression of ZmES22 Influence Expression Profiles of Numerous Starch Synthesis Related Genes at 20 DAP Endosperm
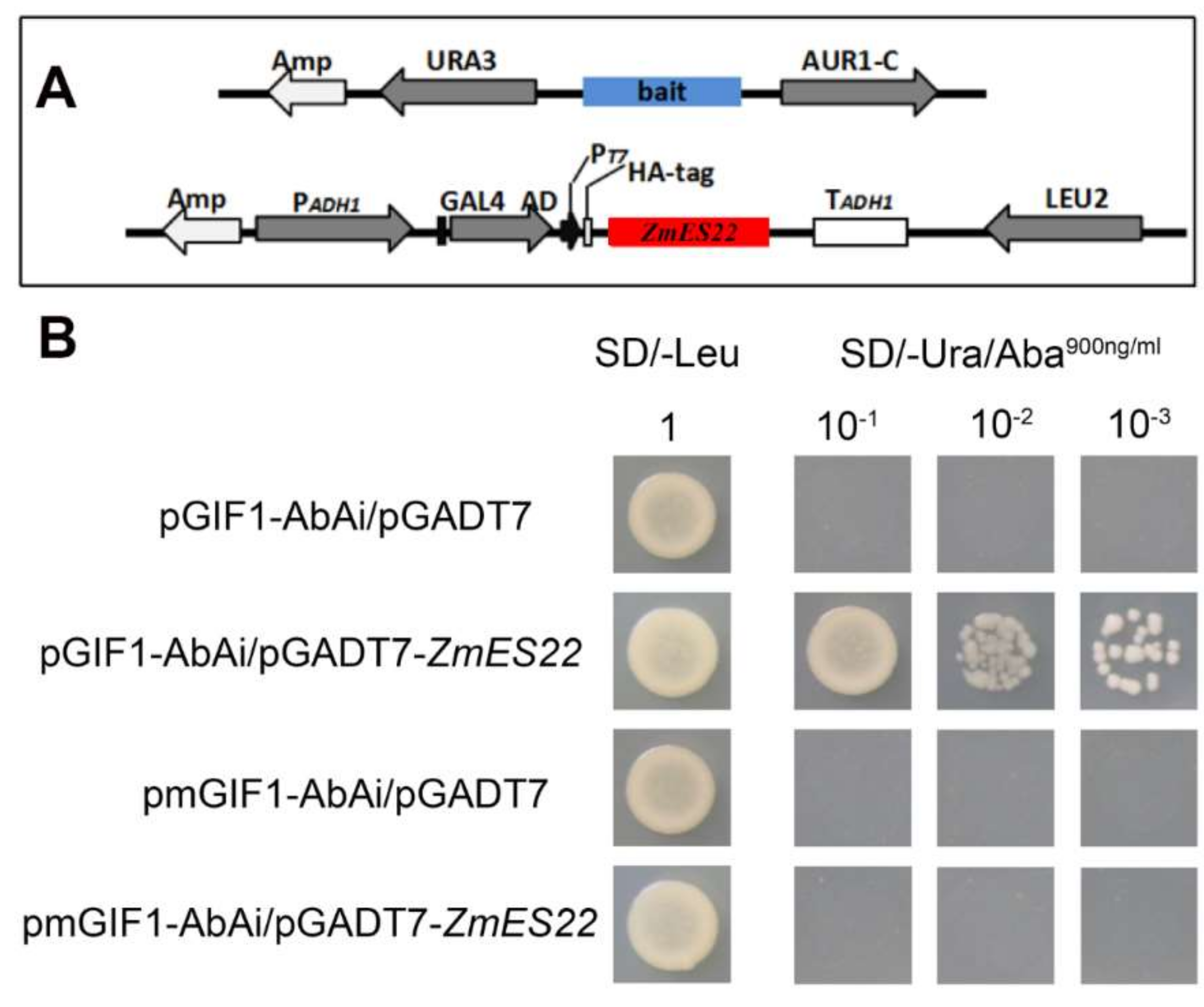
2.6. ZmES22 Could Bind to Promoter GIF1 Gene
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. RNA Extraction and Real-Time RT-PCR Analysis
4.3. Subcellular Localization
4.4. Transcriptional Activation Assay
4.5. Generation of Transgenic Rice Lines
4.6. Determination of Agronomic Characters and Measurement of Grain Quality
4.7. Measurement of Starch Blue Value (BV) and Maximum Absorption Wavelength (kmax)
4.8. Observation of Starch Granules by Scanning Electron Microscopy (SEM)
4.9. RNA-Seq and Data Analysis
4.10. Yeast One-Hybrid Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Qi, X.; Li, S.; Zhu, Y.; Zhao, Q.; Zhu, D.; Yu, J. ZmDof3, a maize endosperm-specific Dof protein gene, regulates starch accumulation and aleurone development in maize endosperm. Plant Mol. Biol. 2017, 93, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y. Towards a Better Understanding of the Metabolic System for Amylopectin Biosynthesis in Plants: Rice Endosperm as a Model Tissue. Plant Cell Physiol. 2002, 43, 718–725. [Google Scholar] [CrossRef] [Green Version]
- James, M.G.; Denyer, K.; Myers, A.M. Starch synthesis in the cereal endosperm. Curr. Opin. Plant Biol. 2003, 6, 215–222. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; Ren, Y.; Qiu, J.; Jiao, G.; Guo, X.; Tang, S.; Wan, J.; Hu, P. OsBT1 encodes an ADP-glucose transporter involved in starch synthesis and compound granule formation in rice endosperm. Sci. Rep. 2017, 7, 40124. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-K.; Hwang, S.-K.; Han, M.; Eom, J.-S.; Kang, H.-G.; Han, Y.; Choi, S.-B.; Cho, M.-H.; Bhoo, S.H.; An, G.; et al. Identification of the ADP-glucose pyrophosphorylase isoforms essential for starch synthesis in the leaf and seed endosperm of rice (Oryza sativa L.). Plant Mol. Biol. 2007, 65, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wang, J.; Zhu, X.; Hao, W.; Wang, L.; Li, Q.; Zhang, L.; He, W.; Lu, B.; Lin, H.; et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef]
- Wang, J.-C.; Xu, H.; Zhu, Y.; Liu, Q.-Q.; Cai, X.-L. OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. J. Exp. Bot. 2013, 64, 3453–3466. [Google Scholar] [CrossRef] [Green Version]
- Sun, C. A Novel WRKY Transcription Factor, SUSIBA2, Participates in Sugar Signaling in Barley by Binding to the Sugar-Responsive Elements of the iso1 Promoter. Plant Cell Online 2003, 15, 2076–2092. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Schippers, J.H.M.; Mieulet, D.; Watanabe, M.; Hoefgen, R.; Guiderdoni, E.; Mueller-Roeber, B. SALT-RESPONSIVE ERF1 Is a Negative Regulator of Grain Filling and Gibberellin-Mediated Seedling Establishment in Rice. Mol. Plant 2014, 7, 404–421. [Google Scholar] [CrossRef]
- Chen, J.; Yi, Q.; Cao, Y.; Wei, B.; Zheng, L.; Xiao, Q.; Xie, Y.; Gu, Y.; Li, Y.; Huang, H.; et al. ZmbZIP91 regulates expression of starch synthesis-related genes by binding to ACTCAT elements in their promoters. J. Exp. Bot. 2016, 67, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zeng, B.; Zhao, H.; Zhang, M.; Xie, S.; Lai, J. Genome-wide Transcription Factor Gene Prediction and their Expressional Tissue-Specificities in Maize. J. Integr. Plant Biol. 2012, 54, 616–630. [Google Scholar] [CrossRef]
- Cai, H.; Chen, Y.; Zhang, M.; Cai, R.; Cheng, B.; Ma, Q.; Zhao, Y. A novel GRAS transcription factor, ZmGRAS20, regulates starch biosynthesis in rice endosperm. Physiol. Mol. Biol. Plants 2017, 23, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Gramzow, L.; Ritz, M.S.; Theißen, G. On the origin of MADS-domain transcription factors. Trends Genet. 2010, 26, 149–153. [Google Scholar] [CrossRef]
- Ma, H.; Yanofsky, M.F.; Meyerowitz, E.M. AGL1-AGL6, an Arabidopsis gene family with similarity to floral homeotic and transcription factor genes. Genes Dev. 1991, 5, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Hizukuri, S.; Juliano, B.O. Purification and structure of amylose from rice starch. Carbohydr. Res. 1986, 148, 299–308. [Google Scholar] [CrossRef]
- Chen, X.; Guo, L.; Du, X.; Chen, P.; Ji, Y.; Hao, H.; Xu, X. Investigation of glycerol concentration on corn starch morphologies and gelatinization behaviours during heat treatment. Carbohydr. Polym. 2017, 176, 56–64. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; de Peer, Y.V.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2001, 30, 325–327. [Google Scholar] [CrossRef]
- Qiao, Z.; Qi, W.; Wang, Q.; Feng, Y.; Yang, Q.; Zhang, N.; Wang, S.; Tang, Y.; Song, R. ZmMADS47 Regulates Zein Gene Transcription through Interaction with Opaque2. PLoS Genet. 2016, 12, e1005991. [Google Scholar] [CrossRef]
- Deschamps, P.; Colleoni, C.; Nakamura, Y.; Suzuki, E.; Putaux, J.-L.; Buleon, A.; Haebel, S.; Ritte, G.; Steup, M.; Falcon, L.I.; et al. Metabolic Symbiosis and the Birth of the Plant Kingdom. Mol. Biol. Evol. 2008, 25, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Xu, S.; Zhang, Z.; Chen, G.; Zhong, Y.; Liu, L.; Zhang, R.; Xue, J.; Guo, D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci. Rep. 2018, 8, 12736. [Google Scholar] [CrossRef]
- Nelson, O.; Pan, D. Starch Synthesis in Maize Endosperms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1995, 46, 475–496. [Google Scholar] [CrossRef]
- Zhai, R.; Feng, Y.; Wang, H.; Zhan, X.; Shen, X.; Wu, W.; Zhang, Y.; Chen, D.; Dai, G.; Yang, Z.; et al. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genom. 2013, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Zhang, Q. Optimising the tissue culture conditions for high efficiency transformation of indica rice. Plant Cell Rep. 2005, 23, 540–547. [Google Scholar] [CrossRef]
- Fu, F.-F.; Xue, H.-W. Coexpression Analysis Identifies Rice Starch Regulator1, a Rice AP2/EREBP Family Transcription Factor, as a Novel Rice Starch Biosynthesis Regulator. Plant Physiol. 2010, 154, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.; Iwamoto, M.; Abe, T.; et al. Rice Annotation Project Database (RAP-DB): An Integrative and Interactive Database for Rice Genomics. Plant Cell Physiol. 2013, 54, e6. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Reads | Mapped Reads | Clean Base (Gb) | Mapped Base (Gb) | Mapping Rate (%) | Concordant Pair Rate (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| WT-1 | 66,417,130 | 61,285,695 | 6.64 | 6.13 | 92.27 | 85.4 | 94.03 | 56.95 |
| WT-2 | 65,482,702 | 60,254,372 | 6.55 | 6.03 | 92.02 | 84.7 | 94.36 | 57.31 |
| ZmES22-1 | 65,615,264 | 60,672,105 | 6.56 | 6.07 | 92.47 | 85.7 | 94.15 | 57.22 |
| ZmES22-2 | 65,985,744 | 60,618,187 | 6.6 | 6.06 | 91.9 | 84.4 | 92.45 | 57.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zha, K.; Xie, H.; Ge, M.; Wang, Z.; Wang, Y.; Si, W.; Gu, L. Expression of Maize MADS Transcription Factor ZmES22 Negatively Modulates Starch Accumulation in Rice Endosperm. Int. J. Mol. Sci. 2019, 20, 483. https://doi.org/10.3390/ijms20030483
Zha K, Xie H, Ge M, Wang Z, Wang Y, Si W, Gu L. Expression of Maize MADS Transcription Factor ZmES22 Negatively Modulates Starch Accumulation in Rice Endosperm. International Journal of Molecular Sciences. 2019; 20(3):483. https://doi.org/10.3390/ijms20030483
Chicago/Turabian StyleZha, Kangyong, Haoxun Xie, Min Ge, Zimeng Wang, Yu Wang, Weina Si, and Longjiang Gu. 2019. "Expression of Maize MADS Transcription Factor ZmES22 Negatively Modulates Starch Accumulation in Rice Endosperm" International Journal of Molecular Sciences 20, no. 3: 483. https://doi.org/10.3390/ijms20030483
APA StyleZha, K., Xie, H., Ge, M., Wang, Z., Wang, Y., Si, W., & Gu, L. (2019). Expression of Maize MADS Transcription Factor ZmES22 Negatively Modulates Starch Accumulation in Rice Endosperm. International Journal of Molecular Sciences, 20(3), 483. https://doi.org/10.3390/ijms20030483