The Link Between Inflammaging and Degenerative Joint Diseases

 , ,
, , 

 and
and
Abstract
:
1. Introduction
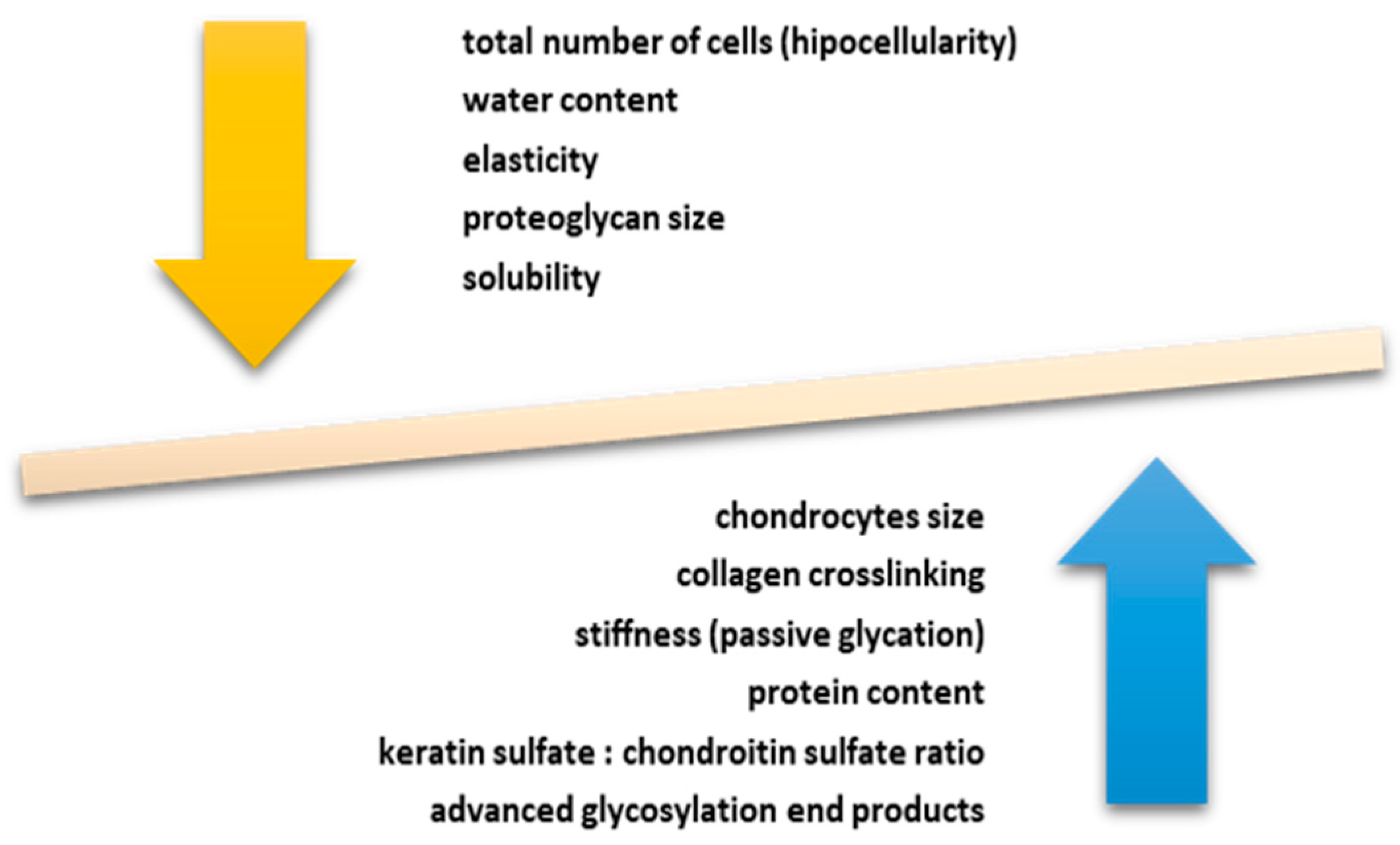
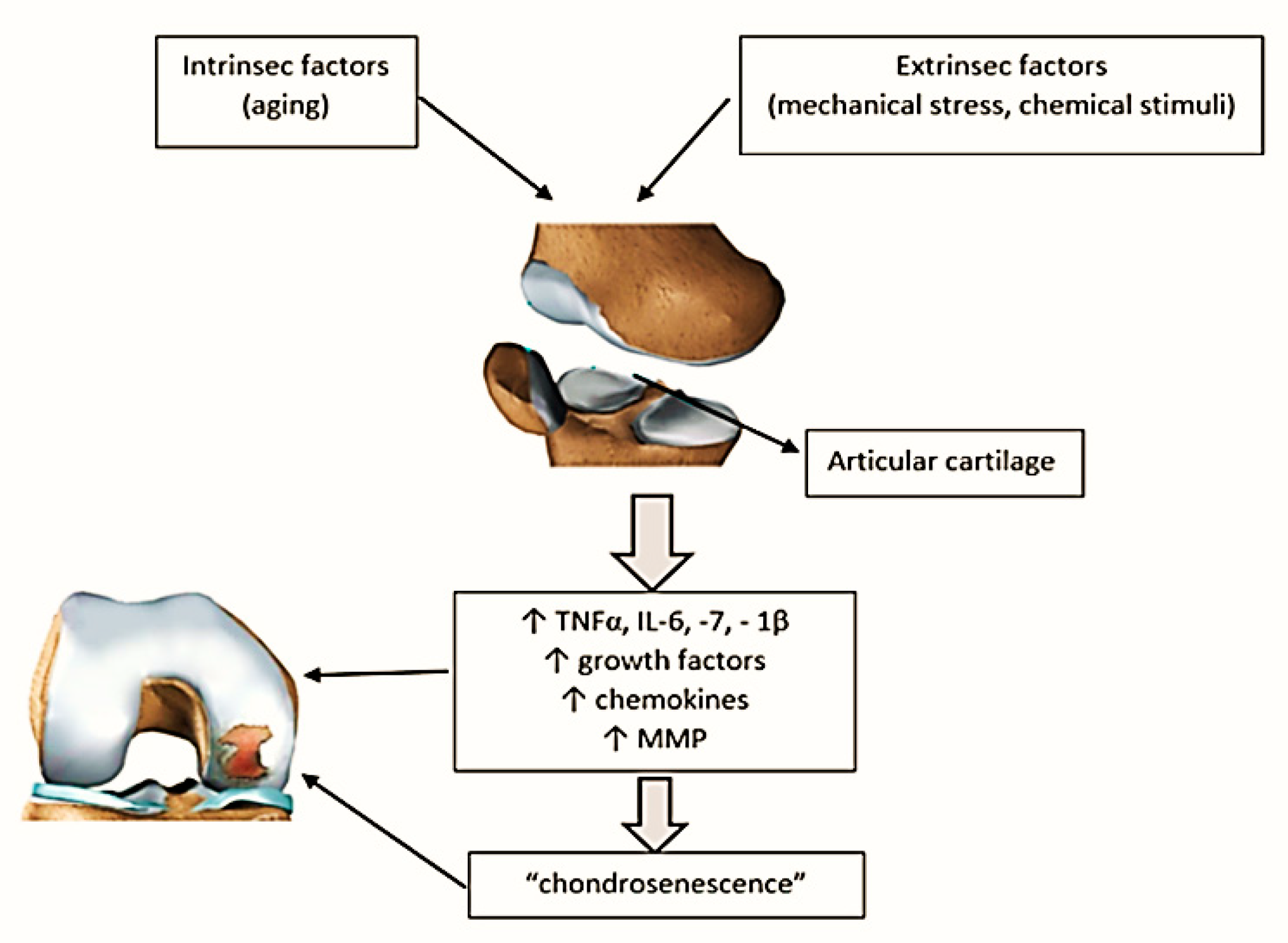
2. The Link between Aging and Articular Cartilage
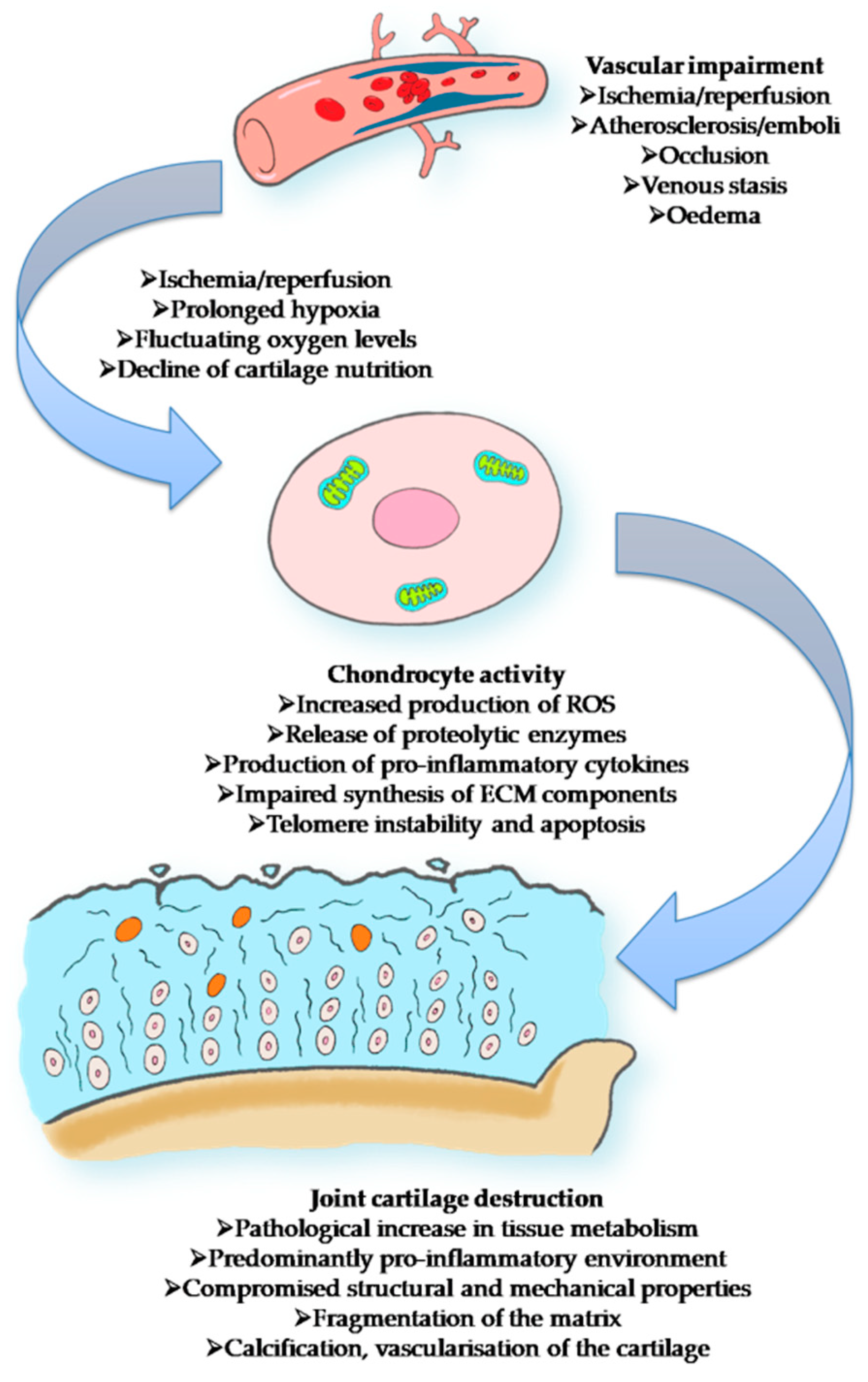
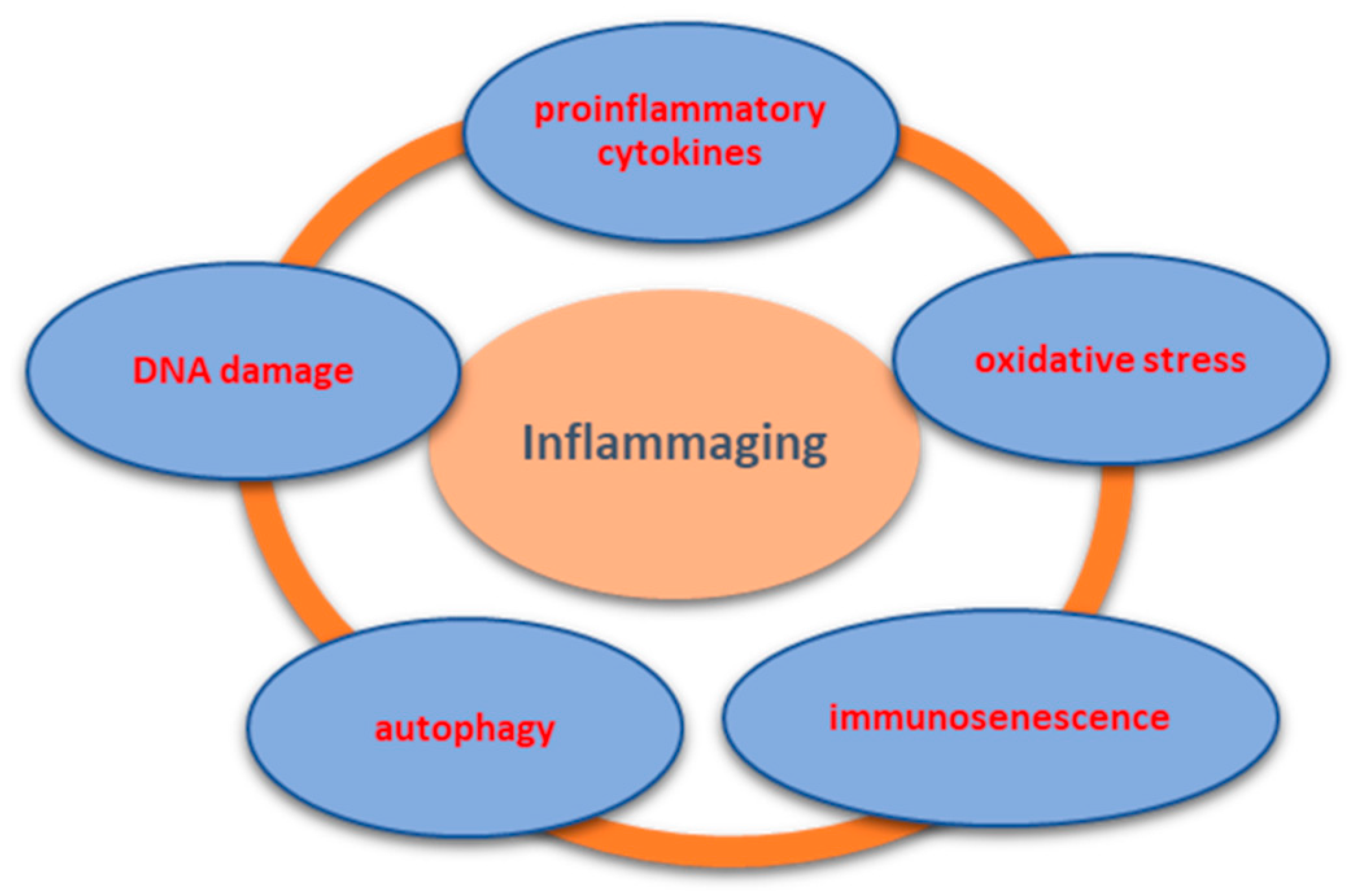
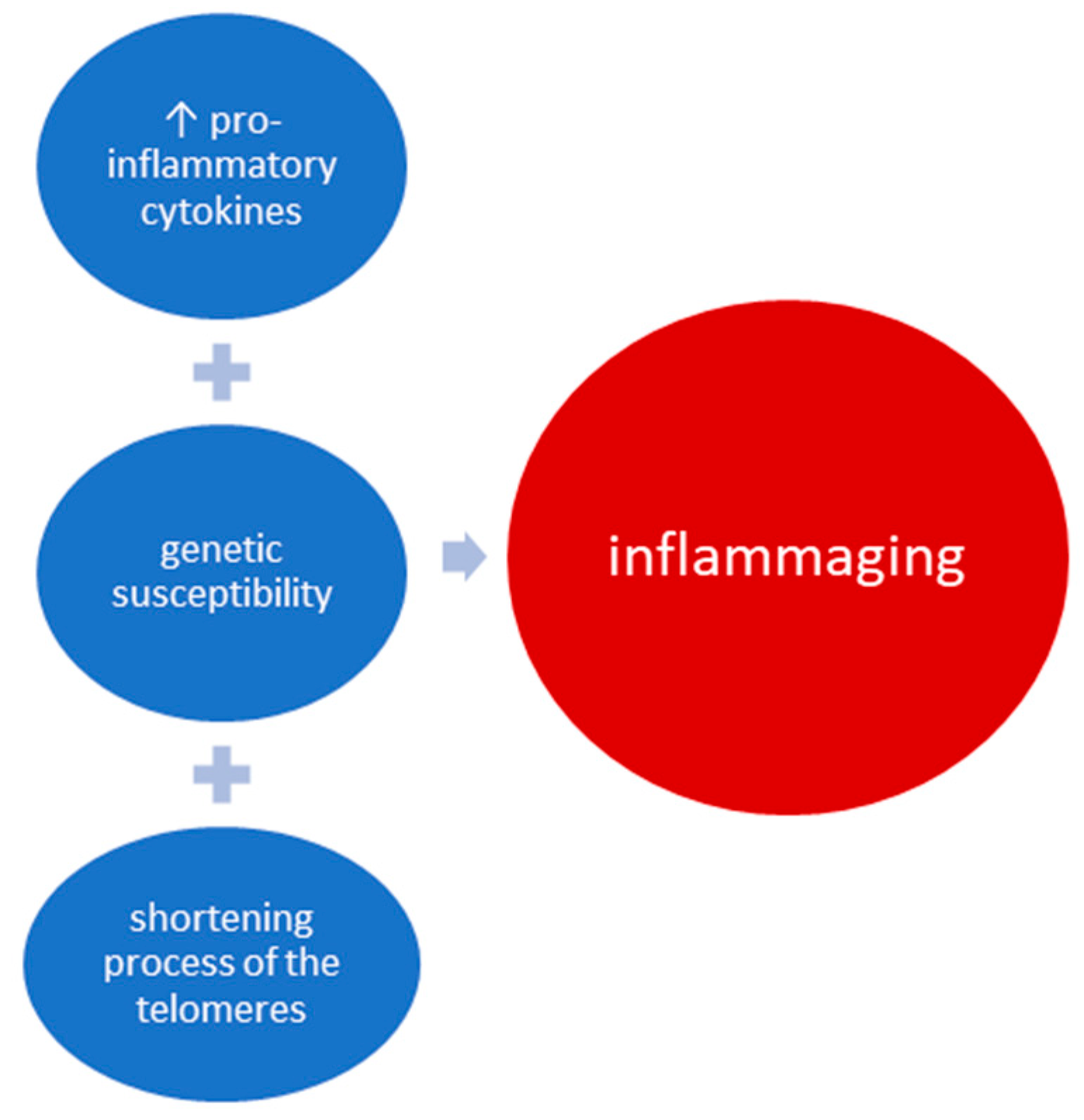
3. Mechanism of Inflammaging and Implications in OA
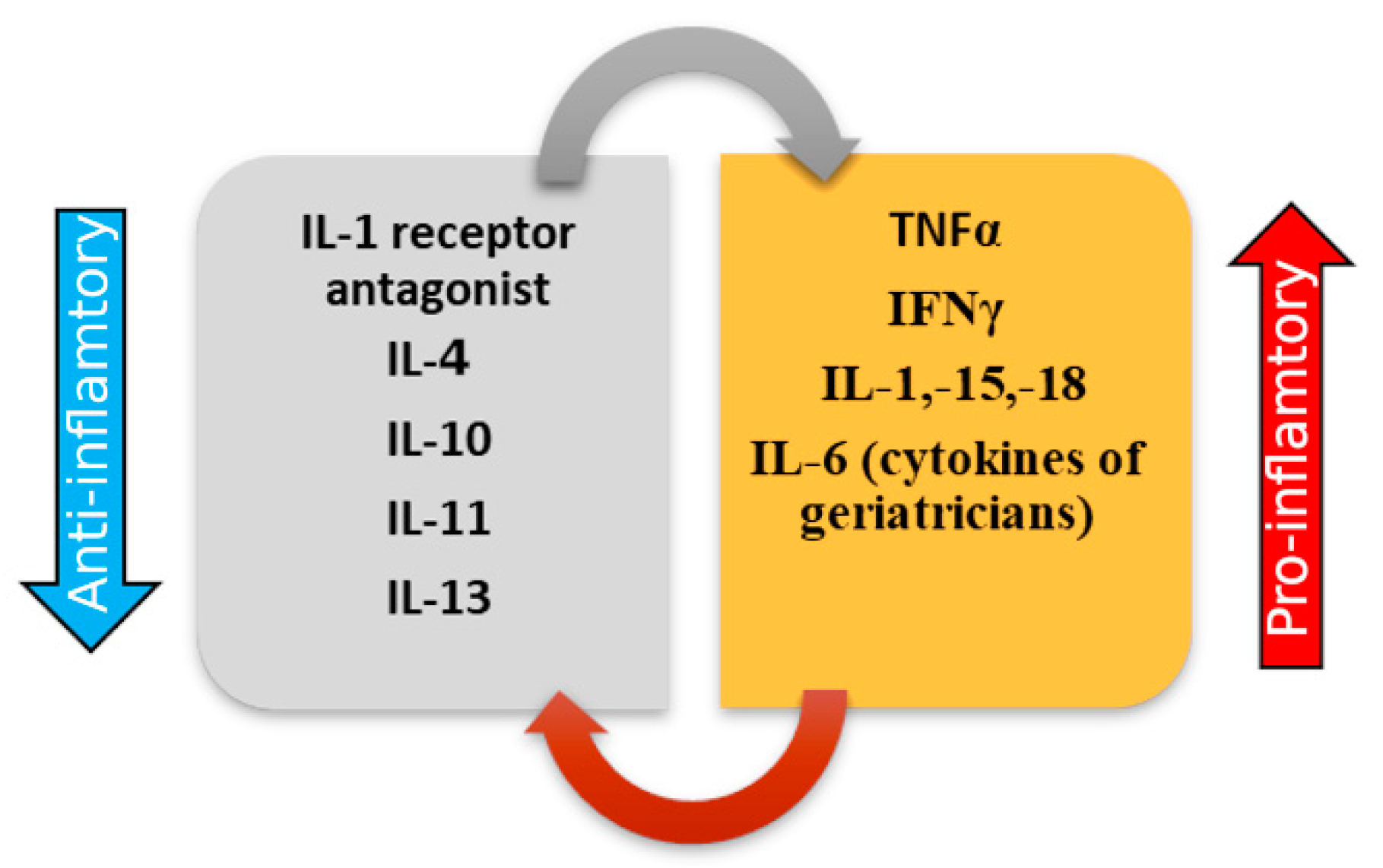
3.1. ProInflammatory Cytokines
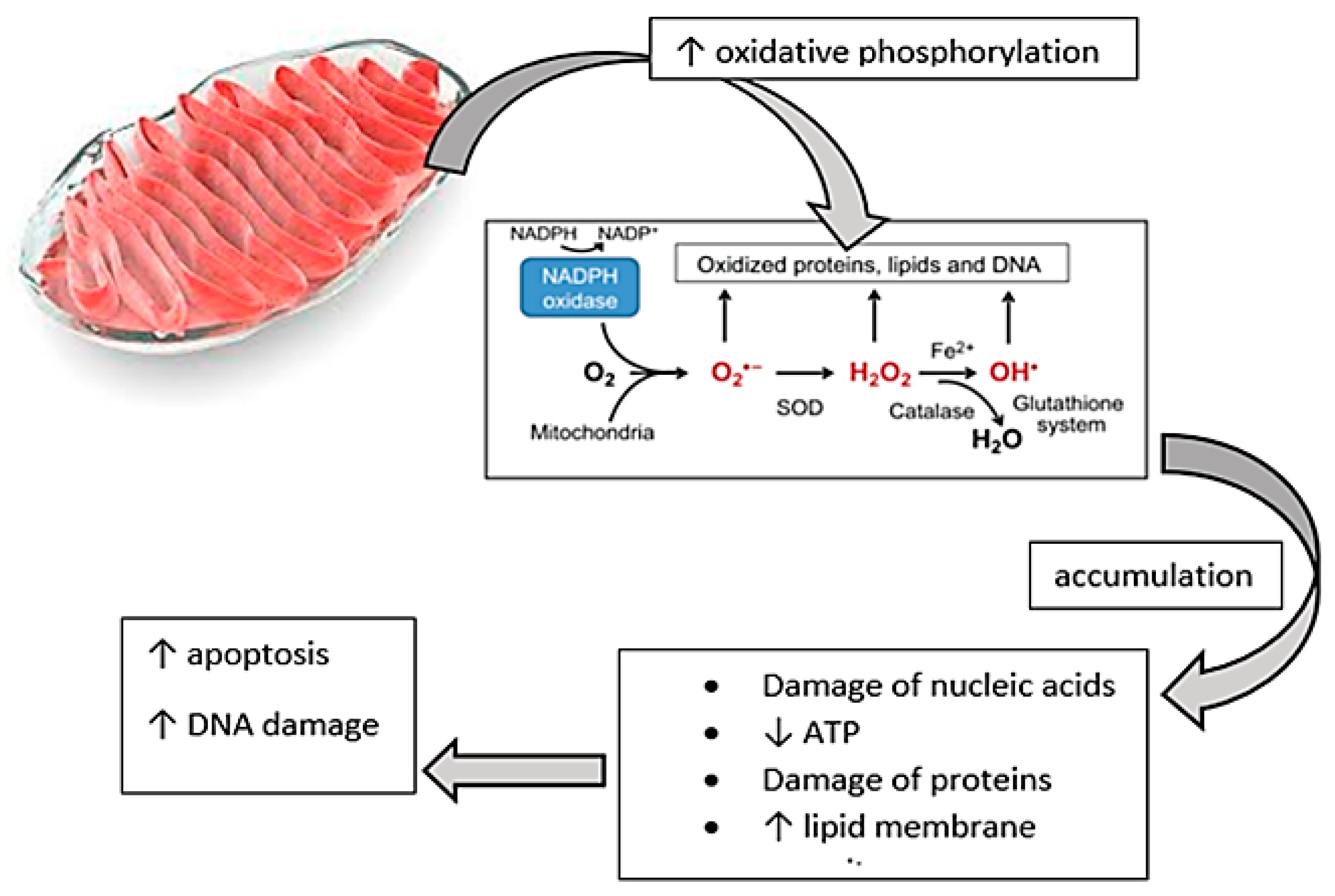
3.2. Oxidative Stress
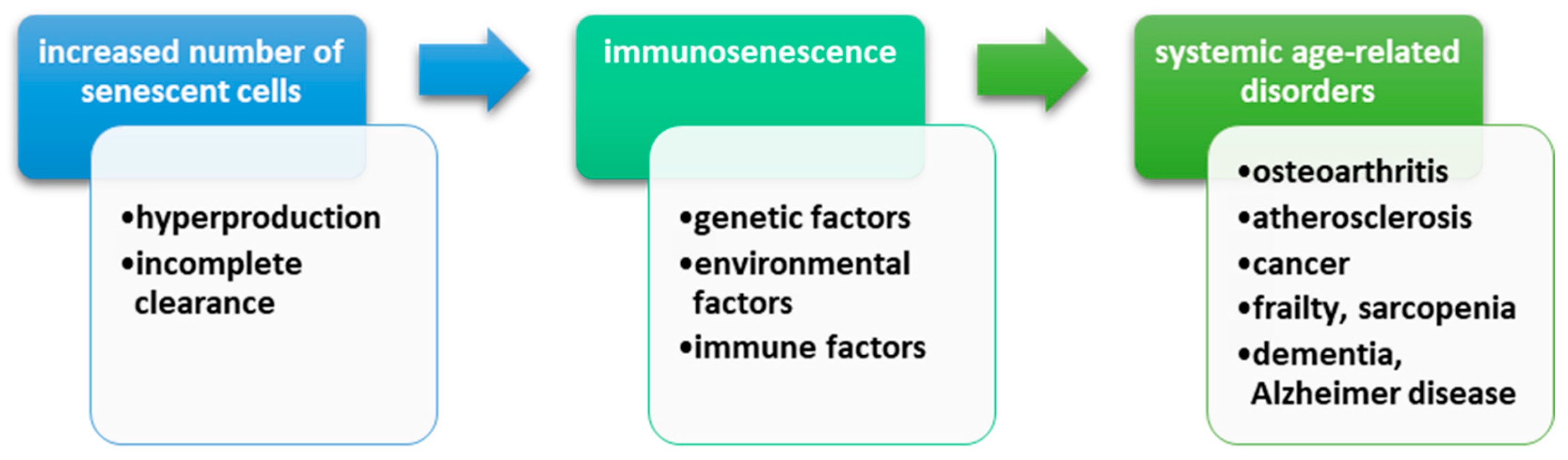
3.3. Immunosenescence and DNA Damage
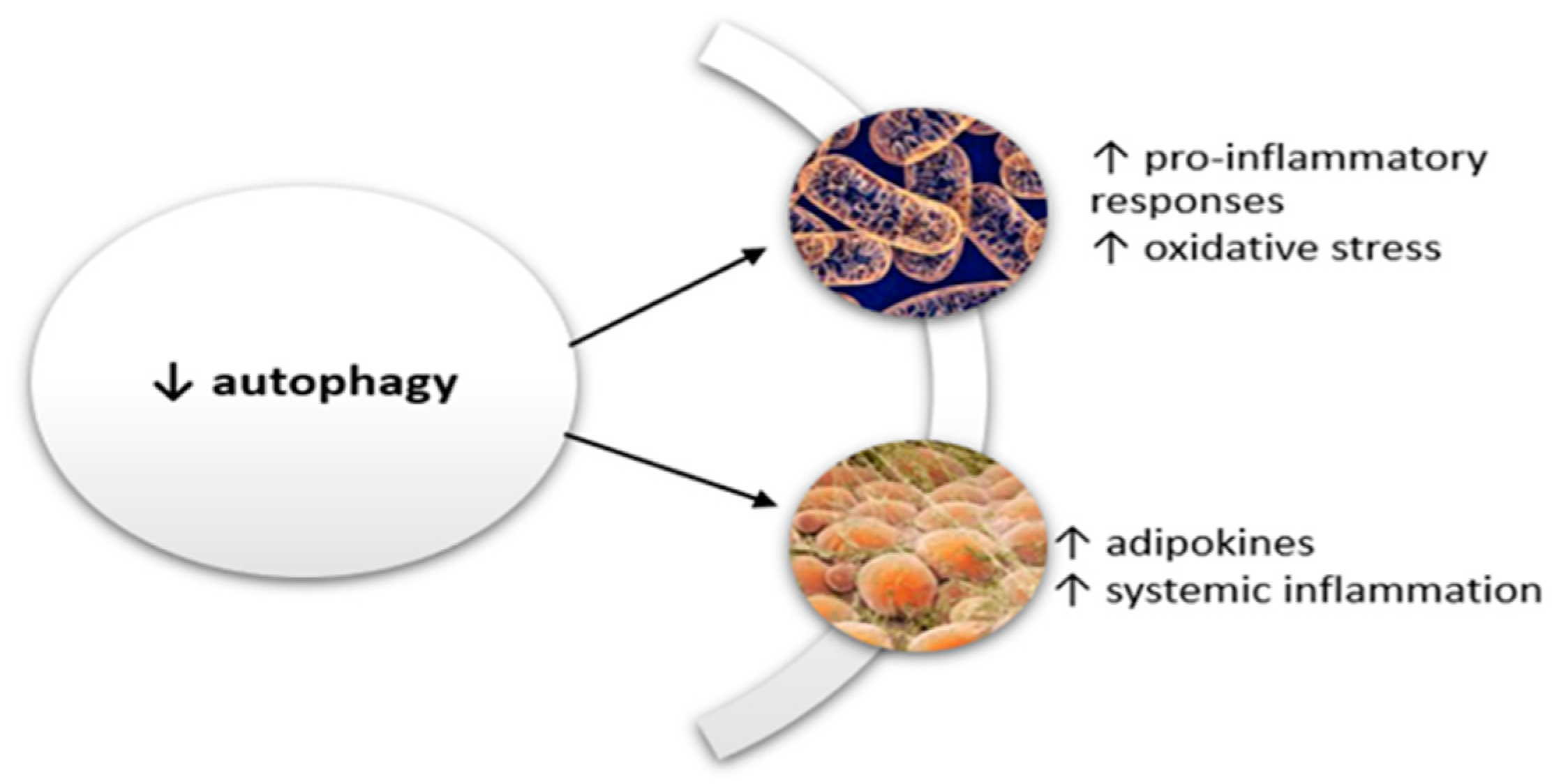
3.4. Autophagy
3.5. Cellular Apoptosis
4. Conclusions
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| TNF α | Tumor necrosis factor alpha |
| IFN γ | Interferon gamma |
| IL | Interleukins |
| CRP | C reactive protein |
| DNA | Dezoxiribonucleic acid |
| ATP | Adenosine triphosphate |
| RNA | Ribonucleic acid |
| OA | Osteoarthritis |
| MMP | Matrix metalloproteinase |
| PDGF | Platelet derived growth factor |
| TGF-β | Transforming growth factor beta |
| IGF-I | Insulin-like growth factor-I |
| BMP-6 | Morphogenic protein 6 |
| p16INK4a | Cyclic-dependent kinase inhibitor |
| MAC | Membrane attack complex |
| SMACs | Second mitochondria-derived activator of caspases |
| IAPs | Inhibitors of apoptosis |
| Apaf-1 | Apoptotic protease activating factor-1 |
| DISC | Death inducing signaling complex |
| FasL | Fas ligand |
References
- Bucci, L.; Ostan, R.; Capri, S.; Salvioli, S.; Cevenini, E.; Celani, L.; Monti, D.; Franceschi, C. Inflamm-Aging: Handbook on Immunosenesce—basic Understanding and Clinical Applications 1; Fulop, T., Franceschi, C., Hirokawa, K., Pawelec, G., Eds.; Springer Science Business Media, B.V.: Dordrecht, The Netherlands, 2009; pp. 893–918. [Google Scholar]
- Rezus, E.; Floria, M.; Grigoriu, A.; Tamba, B.; Rezus, C. Cardiovascular Risk Factors in Chronic Inflammatory Rheumatic Diseases: Modern Assessment and Diagnosis. Curr. Vasc. Pharmacol. 2015, 13, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.T.; Casciaro, M.; Gamgemi, S.; Buquicchio, R. Immunosenescence in aging: Between immune cells depletion and cytokines up-regulation. Clin. Mol. Allergy 2017, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.E.; Yasuhara, R.; Iwamoto, M.; Enomoto-Iwamoto, M. Resident mesenchymal progenitors of articular cartilage. Matrix Biol. 2014, 39, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Cha, B.H.; Kim, J.S.; Ahn, J.; Han, I.; Park, H.; Lee, S.H. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthr. Cartil. 2016, 24, 196–205. [Google Scholar] [CrossRef]
- Brandl, A.; Hartmann, A.; Bechmann, V.; Graf, B.; Nerlich, M.; Angele, P. Oxidative stress induces senescence in chondrocytes. J. Orthop. Res. 2011, 29, 111–1120. [Google Scholar] [CrossRef]
- Dillon, C.F.; Rasch, E.K.; Gu, Q.; Hirsch, R. Prevalence of knee osteoarthritis in the United States: Arthritis data from the Third National Health and Nutrition Examination Survey. J. Rheumatol. 2006, 33, 2271–2279. [Google Scholar]
- Mobasheri, A.; Matta, C.; Zákány, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef]
- Findlay, D.M. Vascular pathology and osteoarthritis. Rheumatology 2007, 46, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- Altindag, O.; Erel, O.; Aksoy, N.; Selek, S.; Celik, H.; Karaoglanoglu, M. Increased oxidative stress and its relation with collagen metabolism in knee osteoarthritis. Rheumatol. Int. 2007, 27, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta (BBA) 2016, 1862, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; van Trieu, N.; Nakamura, H.; Hongo-Masuko, K.; Kato, T.; Nishioka, K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: Oxidative stress induces chondrocyte telomere instability and down regulation of chondrocyte function. Arthritis Res. Ther. 2005, 7, R380. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage–bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632. [Google Scholar] [CrossRef] [PubMed]
- Ershler, W.B. Interleukin-6: A cytokine for gerontologists. J. Am. Geriatr. Soc. 1993, 41, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Bild, V.; Ababei, D.C.; Neamtu, M.; Vasincu, A.; Bild, W.; Stanciu, G.D.; Tamba, B.I.; Solcan, G.; Beschea Chiriac, S. Isobolar analysis of the binary fixed-ratio combination of acetylsalicilic acid-acetaminophen. Farmacia 2017, 65, 563–566. [Google Scholar]
- Bruunsgaard, H. Effects of tumor necrosis factor-alpha and interleukin-6 in elderly populations. Eur. Cytokine Netw. 2002, 13, 389–391. [Google Scholar] [PubMed]
- Spector, T.D.; Hart, D.J.; Nandra, D.; Doyle, D.V.; Mackillop, N.; Gallimore, J.R.; Pepys, M.B. Low-level increases in serum C-reactive protein are present in early osteoarthritis of the knee and predict progressive disease. Arthritis Rheum. 1997, 40, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Livshits, G.; Zhai, G.; Hart, D.J.; Kato, B.S.; Wang, H.; Williams, F.M.; Spector, T.D. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: The Chingford study. Arthritis Rheum. 2009, 60, 2037–2045. [Google Scholar] [CrossRef] [Green Version]
- Penninx, B.W.; Abbas, H.; Ambrosius, W.; Nicklas, B.J.; Davis, C.; Messier, S.P.; Pahor, M. Inflammatory markers and physical function among older adults with knee osteoarthritis. J. Rheumatol. 2004, 31, 2027–2031. [Google Scholar]
- Iurea (Raţă), D.M.; Popa, M.; Chailan, J.F.; Tamba, B.I.; Tudorancea, I.; Peptu, C.A. Ibuprofen-loaded chitosan/poly (maleic anhydride-alt-vinyl acetate) submicronic capsules for pain treatment. J. Bioact. Compat. Polym. 2013, 28, 368–384. [Google Scholar] [CrossRef]
- Alexa, T.; Marza, A.; Voloseniuc, T.; Tamba, B. Enhanced analgesic effects of tramadol and common trace element coadministration in mice: Coadministration of Tramadol and Trace Elements in Mice. J. Neurosci. Res. 2015, 93, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Uritu, C.M.; Mihai, C.T.; Stanciu, G.D.; Dodi, G.; Alexa-Stratulat, T.; Luca, A.; Leon-Constantin, M.M.; Stefanescu, R.; Bild, V.; Melnic, S.; et al. Medicinal plants of the family Lamiaceae in pain therapy: A review. Pain Res. Manag. 2018, 2018, 7801543. [Google Scholar] [CrossRef] [PubMed]
- Tamba, B.I.; Petreus, T.; Constantin, M.M.L.; Rezus, C.; Floria, M.; Rezus, E. Heavy metal trace elements induced antinociception in an experimental mouse model. Rev. Chim. 2015, 66, 976–982. [Google Scholar]
- Stannus, O.P.; Jones, G.; Blizzard, L.; Cicuttini, F.M.; Ding, C. Associations between serum levels of inflammatory markers and change in knee pain over 5 years in older adults: A prospective cohort study. Ann. Rheum. Dis. 2013, 72, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Blake, S.; Song, X.Y.; Lark, M.; Loeser, R.F. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res. Ther. 2008, 10, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.J.; Loeser, R.F., Jr. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 1118–1124. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cellnonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Wang, Q.; Rozelle, A.L.; Lepus, C.M.; Scanzello, C.R.; Song, J.J.; Larsen, D.M.; Crish, J.F.; Bebek, G.; Ritter, S.Y.; Lindstrom, T.M. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011, 17, 1674–1679. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Cellular senescence: Putting the paradoxes in perspective. Curr. Opin. Genet. Dev. 2011, 21, 107–112. [Google Scholar] [CrossRef]
- Price, J.S.; Waters, J.G.; Darrah, C.; Clark, I.M. The role of chondrocyte senescence in osteoarthritis. Aging Cell 2002, 1, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haulica, I.; Bild, W.; Mihaila, C.; Serban, D.N.; Serban, L.; Boisteanu, D.; Ionita, T.; Radasanu, O. Comparative study of the inhibitory effects of adrenomedullin on angiotensin II contraction in rat conductance and resistance arteries. J. Renin. Angiotensin Aldosterone Syst. 2004, 5, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.W.; Lou, S.Q.; Zhang, K. Recovery of function in osteoarthritic chondrocytes induced by p16INK4a-specific siRNA in vitro. Rheumatology 2004, 43, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G.; Aviv, A.; Hunter, D.J.; Hart, D.J.; Gardner, J.P.; Kimura, M.; Lu, X.; Valdes, A.M.; Spector, T.D. Reduction of leucocyte telomere length in radiographic hand osteoarthritis: A population-based study. Ann. Rheum. Dis. 2006, 65, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Kurz, B.; Lemke, A.K.; Fay, J.; Pufe, T.; Grodzinsky, A.J.; Schünke, M. Pathomechanisms of cartilage destruction by mechanical injury. Ann. Anat. 2005, 187, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Mouritzen, U.; Christgau, S.; Lehmann, H.J.; Tanko, L.B.; Christiansen, C. Cartilage turnover assessed with a newly developed assay measuring collagen type II degradation products: Influence of age, sex, menopause, hormone replacement therapy, and body mass index. Ann. Rheum. Dis. 2003, 62, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Guerne, P.A.; Blanco, F.; Kaelin, A.; Desgeorges, A.; Lotz, M. Growth factor responsiveness of human articular chondrocytes in aging and development. Arthritis Rheumol. 1995, 38, 960–968. [Google Scholar] [CrossRef] [Green Version]
- Bobacz, K.; Gruber, R.; Soleiman, A.; Erlacher, L.; Smolen, J.S.; Graninger, W.B. Expression of bone morphogenetic protein 6 in healthy and osteoarthritic human articular chondrocytes and stimulation of matrix synthesis in vitro. Arthritis Rheum. 2003, 48, 2501–2508. [Google Scholar] [CrossRef] [Green Version]
- Yabluchanskiy, A.; Ungvari, Z.; Csiszar, A.; Tarantini, S. Advances and challenges in geroscience research: An update. Physiol. Int. 2018, 105, 298–308. [Google Scholar] [CrossRef]
- Morrisette-Thomas, V.; Cohen, A.A.; Fulop, T.; Riesco, E.; Legault, V.; Li, Q.; Milot, E.; Dusseault-Bélanger, F.; Ferrucci, L. Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech. Ageing Dev. 2014, 139, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.; Newman, A.B. Inflammatory markers in population studies of aging. Ageing Res. Rev. 2011, 10, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Vannucci, S.J.; Bondy, C.A.; Carter, S.D.; Innes, J.F.; Arteaga, M.F.; Trujillo, E.; Ferraz, I.; Shakibaei, M.; Martín-Vasallo, P. Glucose transport and metabolismin chondrocytes: A key to understanding chondrogenesis, skeletal development and cartilage degradation in osteoarthritis. Histol. Histopathol. 2002, 17, 1239–1267. [Google Scholar] [PubMed]
- Salvioli, S.; Capri, M.; Valensin, S.; Tieri, P.; Monti, D.; Ottaviani, E.; Franceschi, C. Inflamm-aging, cytokines and aging: State of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr. Pharm. Des. 2006, 24, 3161–3171. [Google Scholar] [CrossRef]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef]
- De Martinis, M.; Franceschi, C.; Monti, D.; Ginaldi, L. Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 2005, 579, 2035–2039. [Google Scholar] [CrossRef] [Green Version]
- Di Bona, D.; Vasto, S.; Capurso, C.; Christiansen, L.; Deiana, L.; Franceschi, C.; Hurme, M.; Mocchegiani, E.; Rea, M.; Lio, D.; et al. Effect of interleukin-6 polymorphisms on human longevity: A systematic review and meta-analysis. Ageing Res. Rev. 2009, 8, 36–42. [Google Scholar] [CrossRef]
- Lio, D.; Scola, L.; Crivello, A.; Colonna-Romano, G.; Candore, G.; Bonafé, M.; Cavallone, L.; Marchegiani, F.; Olivieri, F.; Franceschi, C.; et al. Inflammation, genetics, and longevity: Further studies on the protective effects in men of IL-10 -1082 promoter SNP and its interaction with TNF-alpha-308 promoter SNP. J. Med. Genet. 2003, 40, 296–299. [Google Scholar]
- Adams, A.A.; Breathnach, C.C.; Katepalli, M.P.; Kohler, K.; Horohov, D.W. Advanced age in horses affects divisional history of T cells and inflammatory cytokine production. Mech. Ageing Dev. 2008, 129, 656–664. [Google Scholar] [CrossRef]
- Evans, W.J.; Paolisso, G.; Abbatecola, A.M.; Corsonello, A.; Bustacchini, S.; Strollo, F.; Lattanzio, F. Frailty and muscle metabolism dysregulation in the elderly. Biogerontology 2010, 11, 527–536. [Google Scholar] [CrossRef]
- Rea, I.M.; Ross, O.A.; Armstrong, M.; McNerlan, S.; Alexander, D.H.; Curran, M.D.; Middleton, D. Interleukin-6-gene C/G 174 polymorphism in nonagenarian and octogenarian subjects in the BELFAST study. Reciprocal effects on IL-6, soluble IL-6 receptor and for IL-10 in serum and monocyte supernatants. Mech. Ageing Dev. 2003, 124, 555–561. [Google Scholar] [CrossRef]
- Lio, D.; Candore, G.; Crivello, A.; Scola, L.; Colonna-Romano, G.; Cavallone, L.; Hoffmann, E.; Caruso, M.; Licastro, F.; Caldarera, C.M.; et al. Opposite effects of interleukin 10 common gene polymorphisms in cardiovascular diseases and in successful ageing: Genetic background of male centenarians is protective against coronary heart disease. J. Med. Genet. 2004, 41, 790–794. [Google Scholar] [PubMed]
- Pes, G.M.; Lio, D.; Carru, C.; Deiana, L.; Baggio, G.; Franceschi, C.; Ferrucci, L.; Oliveri, F.; Scola, L.; Crivello, A.; et al. Association between longevity and cytokine gene polymorphisms. A study in Sardinian centenarians. Aging Clin. Exp. Res. 2004, 16, 244–248. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, A.; Pantell, M.S.; Puterman, E.; Dhabhar, F.S.; Blackburn, E.H.; Yaffe, K.; Cawthon, R.M.; Opresko, P.L.; Hsueh, W.C.; Satterfield, S.; et al. Health Aging and Body Composition Study: Cumulative inflammatory load is associated with short leukocyte telomere length in the Health, Aging and Body Composition Study. PLoS ONE 2011, 6, e19687. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.S.; McGlynn, L.M.; MacIntyre, A.; Johnson, P.C.; Batty, G.D.; Burns, H.; Cavanagh, J.; Deans, K.A.; Ford, I.; McConnachie, A.; et al. Accelerated telomere attrition is associated with relative household income, diet and inflammation in the pSoBid cohort. PLoS ONE 2011, 6, e22521. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Elewaut, D.; McInnes, I.B.; Dayer, J.-M.; Neurath, M.F. How cytokine networks fuel inflammation: Toward a cytokine-based disease taxonomy. Nat. Med. 2013, 19, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Bastiaansen-Jenniskens, Y.; Saris, D.; Creemers, L.B. Pro- and Anti-inflammatory Cytokine Profiles in Osteoarthritis. In Cartilage; Grässel, S., Aszódi, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 81–97. ISBN 978-3-319-45801-4. [Google Scholar]
- Bai, Y.; Gao, S.; Liu, Y.; Jin, S.; Zhang, H.; Su, K. Correlation between Interleukin-17 gene polymorphism and osteoarthritis susceptibility in Han Chinese population. BMC Med. Genet. 2019, 20, 20. [Google Scholar] [CrossRef]
- Vicenti, G.; Bizzoca, D.; Carrozzo, M.; Solarino, G.; Moretti, B. Multi-omics analysis of synovial fluid: A promising approach in the study of osteoarthritis. J. Biol. Regul. Homeost. Agents 2018, 32, 9–13. [Google Scholar]
- Ding, L.; Hong, X.; Sun, B.; Huang, Q.; Wang, X.; Liu, X.; Li, L.; Huang, Z.; Liu, D. IL-37 is associated with osteoarthritis disease activity and suppresses proinflammatory cytokines production in synovial cells. Sci. Rep. 2017, 7, 11601. [Google Scholar] [CrossRef]
- Cannizzo, E.S.; Clement, C.C.; Sahu, R.; Follo, C.; Santambrogio, L. Oxidative stress, inflammaging and immunosenescence. J. Proteom. 2011, 74, 2313–2323. [Google Scholar] [CrossRef]
- Alexa, A.I.; Cantemir, A.; Antioch, I.; Balmus, I.M.; Cojocaru, S.; Luca, A.; Filip, M.A.; Ababei, C.D.; Zamfir, C.L. The Dynamics of the Main Oxidative Stress Chemical Markers in the Serum of Rats Stressed by Various Behavioural Tasks. Rev. Chim. 2017, 68, 350–353. [Google Scholar]
- Ginaldi, L.; De Martinis, M.; Monti, D.; Franceschi, C. The immune system in the elderly: Activation-induced and damage-induced apoptosis. Immunol. Res. 2004, 30, 81–94. [Google Scholar] [CrossRef]
- Marchal, J.; Pifferi, F.; Aujard, F. Resveratrol in mammals: Effects on aging biomarkers, age-related diseases, and life span. Ann. N. Y. Acad. Sci. 2013, 1290, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Draganescu, D.; Ibanescu, C.; Tamba, B.I.; Andritoiu, C.V.; Dodi, G.; Popa, M.I. Flaxseed lignan wound healing formulation: Characterization and in vivo therapeutic evaluation. Int. J. Biol. Macromol. 2015, 72, 614–623. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II. Molecular mechanisms that differentiate apoptosis from programmed necrosis. Toxicol. Pathol. 2013, 41, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Imhof, H.; Sulzbacher, I.; Grampp, S.; Czerny, C.; Youssefzadeh, S.; Kainberger, F. Subchondral bone and cartilage disease: A rediscovered functional unit. Investig. Radiol. 2000, 35, 581–588. [Google Scholar] [CrossRef]
- Hiran, T.S.; Moulton, P.J.; Hancock, J.T. In situ detection of superoxide anions within porcine articular cartilage. Br. J. Biomed. Sci. 1998, 55, 199–203. [Google Scholar]
- Hiran, T.S.; Moulton, P.J.; Hancock, J.T. Detection of superoxide and NADPH oxidase in porcine articular chondrocytes. Free Radic. Biol. Med. 1997, 23, 736–743. [Google Scholar] [CrossRef]
- Clancy, R.M.; Amin, A.R.; Abramson, S.B. The role of nitric oxide in inflammation and immunity. Arthritis Rheum. 1998, 41, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Casagrande, D.; Stains, J.P.; Murthi, A.M. Identification of shoulder osteoarthritis biomarkers: Comparison between shoulders with and without osteoarthritis. J. Shoulder Elbow Surg. 2015, 24, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Balaganur, V.; Pathak, N.N.; Lingaraju, M.C.; More, A.S.; Latief, N.; Kumari, R.R.; Kumar, D.; Tandan, S.K. Effect of Smethylisothiourea, an inducible nitric oxide synthase inhibitor, in joint pain and pathology in surgically induced model of osteoarthritis. Connect. Tissue Res. 2014, 55, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Ramage, L.; Martel, M.-A.; Hardingham, G.E.; Salter, D.M. NMDA receptor expression and activity in osteoarthritic human articular chondrocytes. Osteoarthr. Cartil. 2008, 16, 1576–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokay, N.S.; Yilmaz, I.; Komur, B.; Demiroz, A.S.; Gokce, A.; Devisoglu, S.; Gokay, B.V. A Comparison of the effects of neuronal nitric oxide synthase and inducible nitric oxide synthase inhibition on cartilage damage. BioMed Res. Int. 2016, 2016, 7857345. [Google Scholar] [CrossRef] [PubMed]
- Leonidou, A.; Lepetsos, P.; Mintzas, M.; Kenanidis, E.; Macheras, G.; Tzetis, M.; Potoupnis, M.; Tsiridis, E. Inducible nitric oxide synthase as a target for osteoarthritis treatment. Expert Opin. Ther. Targets 2018, 22, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615. [Google Scholar] [CrossRef]
- Studer, R.; Jaffurs, D.; Stefanovic-Racic, M.; Robbins, P.D.; Evans, C.H. Nitric oxide in osteoarthritis. Osteoarthr. Cartil. 1999, 7, 377–379. [Google Scholar] [CrossRef] [Green Version]
- Stanciu, G.D.; Solcan, G. Acute idiopathic polyradiculoneuritis concurrent with acquired myasthenia gravis in a West Highland white terrier dog. BMC Vet. Res. 2016, 12, 111. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, 4445. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Candore, G.; Accardi, G.; Colonna-Romano, G.; Lio, D. NF-κB pathway activators as potential ageing biomarkers: Targets for new therapeutic strategies. Immun. Ageing 2013, 10, 24. [Google Scholar] [CrossRef]
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruunsgaard, H.; Pedersen, M.; Pedersen, B.K. Aging and proinflammatory cytokines. Curr. Opin. Hematol. 2001, 8, 131–136. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulates the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 2009, 1, 402–411. [Google Scholar] [CrossRef]
- Jones, D.L.; Rando, T.A. Emerging models and paradigms for stem cell ageing. Nat. Cell Biol. 2011, 13, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Cevenini, E.; Monti, D.; Franceschi, C. Inflamm-ageing. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Vasile, E.; Tomita, Y.; Brown, L.F.; Kocher, O.; Dvorak, H.F. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: Evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001, 15, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Albertini, M.C.; Orciani, M.; Ceka, A.; Cricca, M.; Procopio, A.D.; Bonafè, M. DNA damage response (DDR) and senescence: Shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 2015, 6, 35509–35521. [Google Scholar] [CrossRef] [PubMed]
- Bonafè, M.; Storci, G.; Franceschi, C. Inflamm-aging of the stem cell niche: Breast cancer as a paradigmatic example: Breakdown of the multi-shell cytokine network fuels cancer in aged people. Bioessays 2012, 34, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; van den Berg, W.B. Osteoarthritis in the context of ageing and evolution. Loss of chondrocyte differentiation block during ageing. Ageing Res. Rev. 2008, 7, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, D.H.; Sokolove, J.; Sharpe, O.; Erhart, J.C.; Chandra, P.E.; Lahey, L.J.; Erhart, J.C.; Chandra, P.E.; Lahey, L.J.; Lindstrom, T.M.; et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res. Ther. 2012, 14, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clutterbuck, A.L.; Smith, J.R.; Allaway, D.; Harris, P.; Liddell, S.; Mobasheri, A. High throughput proteomic analysis of the secretome in an explant model of articular cartilage inflammation. J. Proteom. 2011, 74, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.; Soder, S.; Skhirtladze, C.; Schmitz, N.; Gebhard, P.M.; SesselmAnn, S.; Aigner, T. DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthr. Cartil. 2012, 20, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [Green Version]
- White, E.; Lowe, S.W. Eating to exit: Autophagy-enabled senescence revealed. Genes Dev. 2009, 23, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Drevon, C.A.; Eckel, J. Secreted proteins from adipose tissue and skeletal muscle—Adipokines, myokines and adipose/muscle cross-talk. Arch. Physiol. Biochem. 2011, 117, 47–56. [Google Scholar] [CrossRef] [PubMed]
- De Heredia, F.P.; Sonia, G.M.; Ascension, M. Chronic and degenerative diseases: Obesity, inflammation and the immune system. Proc. Nutr. Soc. 2012, 71, 332–338. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, H.; Im, G.-I. Autophagy in osteoarthritis. Connect. Tissue Res. 2017, 58, 497–508. [Google Scholar] [CrossRef]
- Li, Y.-S.; Zhang, F.-J.; Zeng, C.; Luo, W.; Xiao, W.-F.; Gao, S.-G.; Lei, G.-H. Autophagy in osteoarthritis. Jt. Bone Spine 2016, 83, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Guo, W.; Chen, M.; Huang, J.; Yuan, Z.; Zhang, Y.; Wang, M.; Li, P.; Peng, J.; Wang, A.; et al. Extracellular Vesicles and Autophagy in Osteoarthritis. BioMed Res. Int. 2016, 2016, 2428915. [Google Scholar] [CrossRef]
- Zhang, Y.; Vasheghani, F.; Li, Y.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.-P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Savill, J.S.; Wyllie, A.H.; Henson, J.E.; Savill, J.S.; Wyllie, A.H.; Henson, J.E.; Walport, M.J.; Henson, P.M.; Haslett, C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Investig. 1989, 83, 865–875. [Google Scholar] [CrossRef]
- Firestein, G.S.; Nguyen, K.; Aupperle, K.R.; Yeo, M.; Boyle, D.L.; Zvaifler, N.J. Apoptosis in rheumatoid arthritis: p53 overexpression in rheumatoid arthritis synovium. Am. J. Pathol. 1996, 149, 2143–2151. [Google Scholar] [PubMed]
- Hasunuma, T.; Kayagaki, N.; Asahara, H.; Motokawa, S.; Kobata, T.; Yagita, H.; Aono, H.; Sumida, T.; Okumura, K.; Nishioka, K. Accumulation of soluble Fas in inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum. 1997, 40, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Sharif, M. Chondrocyte apoptosis: A cause or consequence of osteoarthritis. Int J. Rheum. Dis. 2011, 14, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, S.W.; Helminen, H.J.; Khillan, J.S.; Bao, Y.; Prockop, D.J. Apoptosis of chondrocytes in transgenic mice lacking collagen II. Exp. Cell Res. 1997, 235, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Ochs, R.L.; Komiya, S.; Lotz, M. Linkage of chondrocyte apoptosis and cartilage degradation in human osteoarthritis. Arthritis Rheum. 1998, 41, 1632–1638. [Google Scholar] [CrossRef]
- Roach, H.I.; Aigner, T.; Kouri, J.B. Chondroptosis: A variant of apoptotic cell death in chondrocytes. Apoptosis 2004, 9, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Kim, H.A.; Roach, H.I. Apoptosis in osteoarthritis. Rheum. Dis. Clin. N. Am. 2004, 30, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Suh, D.I.; Song, Y.W. Relationship between chondrocyte apoptosis and matrix depletion in human articular cartilage. J. Rheumatol. 2001, 28, 2038–2045. [Google Scholar]
- Pulai, J.I.; del Carlo, M., Jr.; Loeser, R.F. The α5β1 integrin provides matrix survival signals for normal and osteoarthritic human articular chondrocytes in vitro. Arthritis Rheum. 2002, 46, 1528–1535. [Google Scholar] [CrossRef]
- Saito, S.; Murakoshi, K.; Kotake, S.; Kamatani, N.; Tomatsu, T. Granzyme B induces apoptosis of chondrocytes with natural killer cell-like cytotoxicity in rheumatoid arthritis. J. Rheumatol. 2008, 35, 1932–1943. [Google Scholar]
- Green, D. Means to an End: Apoptosis and other Cell Death Mechanisms; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011; ISBN 978-0-87969-888-1. [Google Scholar]
- Raychaudhuri, S. A minimal model of signaling network elucidates cell-to-cell stochastic variability in apoptosis. PLoS ONE 2010, 5, e11930. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Chapter 18 Apoptosis: Programmed Cell Death Eliminates Unwanted Cells. In Molecular Biology of the Cell (Textbook), 5th ed.; Garland Science: New York, NY, USA, 2008; p. 1032. ISBN 978-0-8153-4105-5. [Google Scholar]
- Gonzalez, D.; Bejarano, I.; Barriga, C.; Rodriguez, A.B.; Pariente, J.A. Oxidative stress-induced caspases are regulated in human myeloid HL-60 cells by calcium signal. Curr. Signal. Transduct. Ther. 2010, 5, 181–186. [Google Scholar] [CrossRef]
- Brüne, B. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ. 2003, 10, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesik, S.W.; Shi, Y.; Structural Biology. Controlling the caspases. Science 2001, 294, 1477–1478. [Google Scholar] [CrossRef] [PubMed]
- Dejean, L.M.; Martinez-Caballero, S.; Kinnally, K.W. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ. 2006, 13, 1387–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejean, L.M.; Martinez-Caballero, S.; Manon, S.; Kinnally, K.W. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim. Biophys. Acta 2006, 1762, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Senescent Chondrocytes | References |
|---|---|
| formation of senescence-associated heterochromatin | [30] |
| Increase of: | |
| expression of cyclin-dependent kinase inhibitor (p16INK4a) | [30,31,32] |
| expression of senescence-associated β-galactosidase | [30,32] |
| telomere length reduction | [34] |
| free oxygen radicals, ↑ oxidative stress | [35] |
| Matrix Metallopeptidase 13 (MMP-13) | [28,36] |
| Decrease of: | |
| transforming growth factor-beta (TGF-β) | [37] |
| platelet derived growth factor (PDGF) | [37] |
| insulin-like growth factor-I (IGF-I) | [37] |
| synthesis of proteoglycans | [38,39] |
| Increase in Oxidative Stress Leads to | |
|---|---|
| Acceleratedcatabolic Processes with Increase in | Slowed Anabolic Processes with Decrease in |
| proinflammatory cytokines | extracellular matrix |
| MMP | growth factor expression |
| accumulation of cellular detrudes | protein synthesis |
| apoptosis | DNA synthesis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezuș, E.; Cardoneanu, A.; Burlui, A.; Luca, A.; Codreanu, C.; Tamba, B.I.; Stanciu, G.-D.; Dima, N.; Bădescu, C.; Rezuș, C. The Link Between Inflammaging and Degenerative Joint Diseases. Int. J. Mol. Sci. 2019, 20, 614. https://doi.org/10.3390/ijms20030614
Rezuș E, Cardoneanu A, Burlui A, Luca A, Codreanu C, Tamba BI, Stanciu G-D, Dima N, Bădescu C, Rezuș C. The Link Between Inflammaging and Degenerative Joint Diseases. International Journal of Molecular Sciences. 2019; 20(3):614. https://doi.org/10.3390/ijms20030614
Chicago/Turabian StyleRezuș, Elena, Anca Cardoneanu, Alexandra Burlui, Andrei Luca, Cătălin Codreanu, Bogdan Ionel Tamba, Gabriela-Dumitrița Stanciu, Nicoleta Dima, Codruța Bădescu, and Ciprian Rezuș. 2019. "The Link Between Inflammaging and Degenerative Joint Diseases" International Journal of Molecular Sciences 20, no. 3: 614. https://doi.org/10.3390/ijms20030614
APA StyleRezuș, E., Cardoneanu, A., Burlui, A., Luca, A., Codreanu, C., Tamba, B. I., Stanciu, G. -D., Dima, N., Bădescu, C., & Rezuș, C. (2019). The Link Between Inflammaging and Degenerative Joint Diseases. International Journal of Molecular Sciences, 20(3), 614. https://doi.org/10.3390/ijms20030614