Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
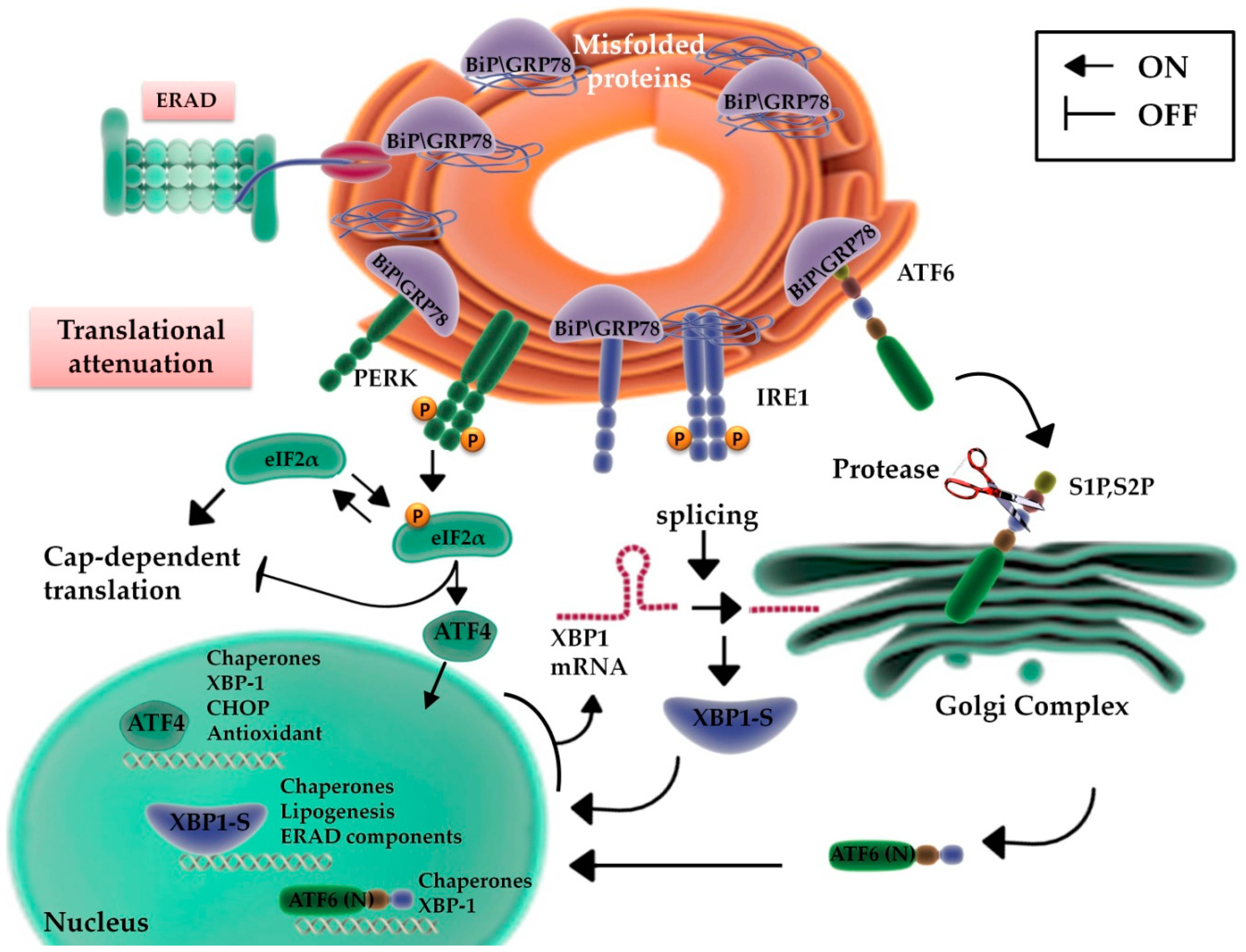
2. ER Function: From Protein Production to UPR
2.1. UPR Signaling
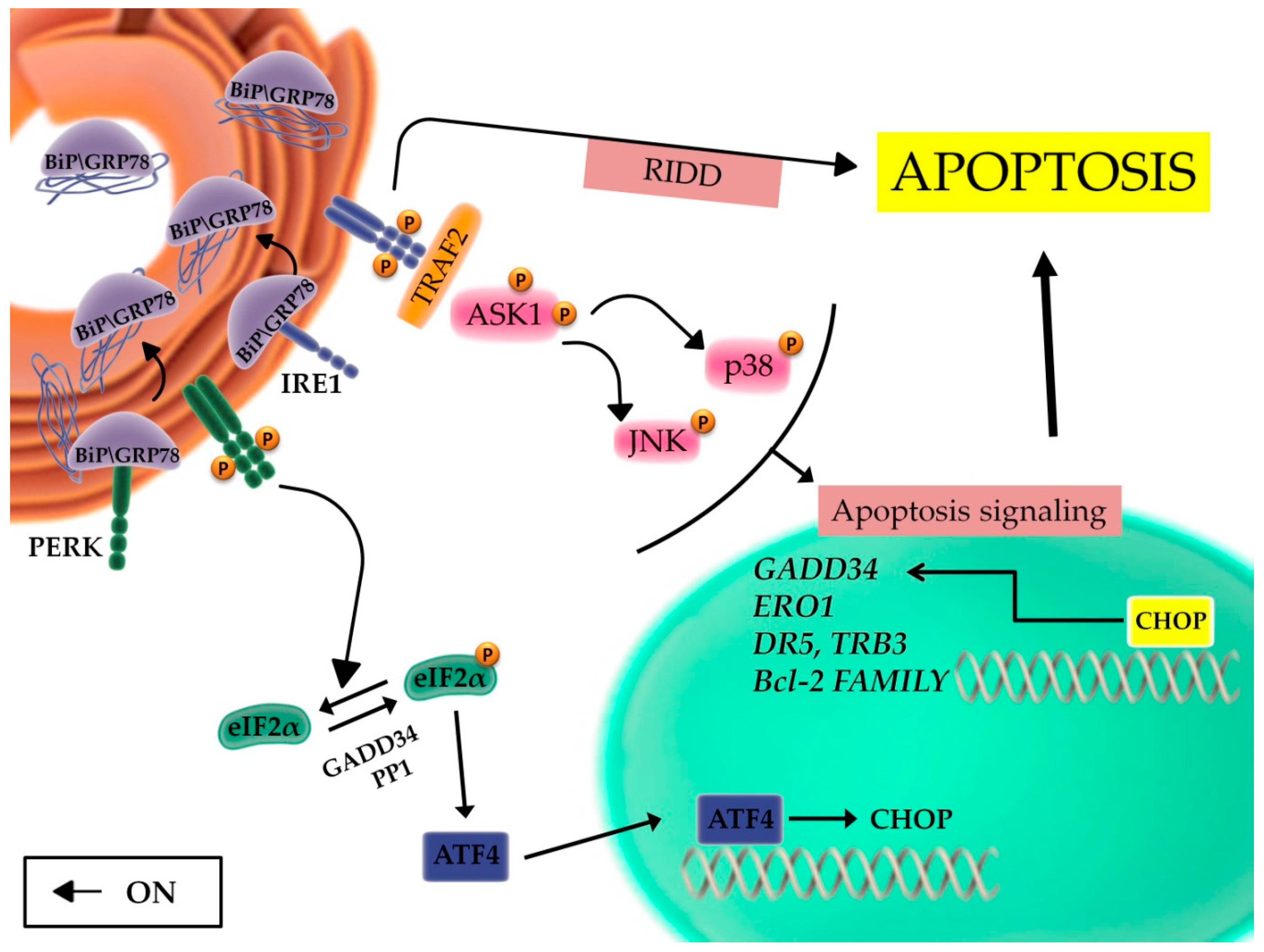
2.2. The Unfolded Protein Response and the Balance between Autophagy, Survival, and Apoptosis
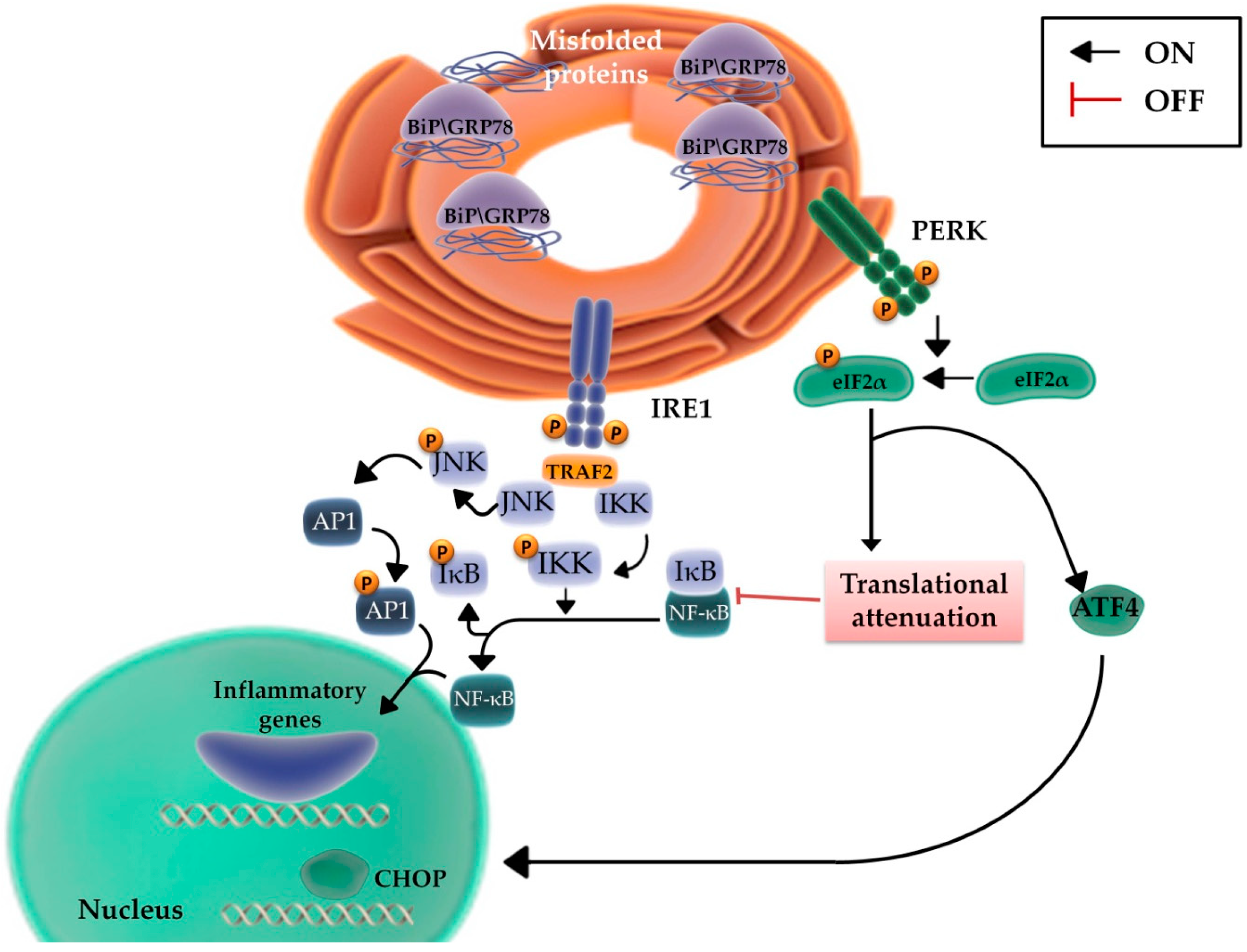
2.3. The Role of the Inflammatory Signaling Cascade during the UPR
3. Endoplasmic Reticulum Stress and UPR in Breast Cancer and Their Involvement in Drug Resistance
3.1. Aberrant UPR Activation in Breast Cancer
3.2. UPR Activation and Drug Resistance in Breast Cancer
4. Molecular Chaperones in Protection from ER Stress and Drug Resistance in BC Cells
4.1. TRAP1 (HSP75)
4.2. HSP90
4.3. HSP27
5. Future Prospective: Autophagy and UPR as Novel Molecular Targets
Funding
Conflicts of Interest
References
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Shajahan, A.N.; Wang, Y.; Tyson, J.J.; Riggins, R.B.; Weiner, L.M.; Bauman, W.T.; Xuan, J.; Zhang, B.; Facey, C.; et al. Endoplasmic reticulum stress, the unfolded protein response, and gene network modeling in antiestrogen resistant breast cancer. Horm. Mol. Biol. Clin. Investig. 2011, 5, 35–44. [Google Scholar] [CrossRef]
- Clarke, R.; Cook, K.L. Unfolding the Role of Stress Response Signaling in Endocrine Resistant Breast Cancers. Front. Oncol. 2015, 5, 140. [Google Scholar] [CrossRef]
- Andruska, N.; Zheng, X.; Yang, X.; Helferich, W.G.; Shapiro, D.J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene 2015, 34, 3760–3769. [Google Scholar] [CrossRef]
- Rajapaksa, G.; Nikolos, F.; Bado, I.; Clarke, R.; Gustafsson, J.A.; Thomas, C. ERbeta decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene 2015, 34, 4130–4141. [Google Scholar] [CrossRef]
- Cook, K.L.; Shajahan, A.N.; Warri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012, 72, 3337–3349. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Cook, K.L.; Clarke, P.A.; Clarke, R. Targeting GRP78 and antiestrogen resistance in breast cancer. Future Med. Chem. 2013, 5, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.L.; Gao, D.S. Endoplasmic reticulum proteins quality control and the unfolded protein response: The regulative mechanism of organisms against stress injuries. Biofactors 2014, 40, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Nagata, K. Hsp47 as a collagen-specific molecular chaperone. Methods Enzymol. 2011, 499, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.; Gauss, R.; Horn, S.C.; Neuber, O.; Sommer, T. The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009, 458, 453–460. [Google Scholar] [CrossRef]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009, 28, F96–F103. [Google Scholar]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Ali, M.M. UPR Signal Activation by Luminal Sensor Domains. Int. J. Mol. Sci. 2013, 14, 6454–6466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Sannino, S.; Brodsky, J.L. Targeting protein quality control pathways in breast cancer. BMC Biol. 2017, 15, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, T.; Simmen, T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Zhang, Q.H.; Lu, Y.J.; Ren, K.; Yi, G.H. Involvement of the IRE1alpha-XBP1 pathway and XBP1s-dependent transcriptional reprogramming in metabolic diseases. DNA Cell Biol. 2015, 34, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef] [PubMed]
- Jain, B.P. An Overview of Unfolded Protein Response Signaling and Its Role in Cancer. Cancer Biother. Radiopharm. 2017, 32, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [PubMed]
- Swart, C.; Du Toit, A.; Loos, B. Autophagy and the invisible line between life and death. Eur. J. Cell Biol. 2016, 95, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Wei, J.; You, L.; Liu, H.; Qian, W. Autophagy regulation and its dual role in blood cancers: A novel target for therapeutic development (Review). Oncol. Rep. 2018, 39, 2473–2481. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Macdonald, D.C.; Ni Chonghaile, T.; Gupta, S.; Samali, A. Bcl-2 family on guard at the ER. Am. J. Physiol. Cell Physiol. 2009, 296, C941–953. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2015, 47, 146–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Ladiges, W.C.; Knoblaugh, S.E.; Morton, J.F.; Korth, M.J.; Sopher, B.L.; Baskin, C.R.; MacAuley, A.; Goodman, A.G.; LeBoeuf, R.C.; Katze, M.G. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 2005, 54, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef]
- Deng, X.; Xiao, L.; Lang, W.; Gao, F.; Ruvolo, P.; May, W.S., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001, 276, 23681–23688. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Shu, C.W.; Xu, W.; Shiau, C.W.; Grant, D.; Vasile, S.; Cosford, N.D.; Reed, J.C. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 2009, 284, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jager, R.; Gorman, A.M.; Samali, A. The Unfolded Protein Response in Breast Cancer. Cancers 2018, 10. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Hermel, E.; Castro-Obregon, S.; del Rio, G.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J. Biol. Chem. 2001, 276, 33869–33874. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Rahman, S.; Jan, A.T.; Archana, A.; Kim, J. Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp. Mol. Med. 2017, 49, e389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gokcen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-kappaB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Baldwin, A.S. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef]
- Kohler, B.A.; Sherman, R.L.; Howlader, N.; Jemal, A.; Ryerson, A.B.; Henry, K.A.; Boscoe, F.P.; Cronin, K.A.; Lake, A.; Noone, A.M.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J. Natl. Cancer Inst. 2015, 107, djv048. [Google Scholar] [CrossRef]
- Feng, Y.X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Syu, J.P.; Chi, J.T.; Kung, H.N. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; van der Kogel, A.J.; Sweep, F.C.; Span, P.N. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother. Oncol. 2013, 108, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Notte, A.; Rebucci, M.; Fransolet, M.; Roegiers, E.; Genin, M.; Tellier, C.; Watillon, K.; Fattaccioli, A.; Arnould, T.; Michiels, C. Taxol-induced unfolded protein response activation in breast cancer cells exposed to hypoxia: ATF4 activation regulates autophagy and inhibits apoptosis. Int. J. Biochem. Cell Biol. 2015, 62, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, X.A.; Hu, J.; Jiang, J.K.; Li, Y.; Chan-Salis, K.Y.; Gu, Y.; Chen, G.; Thomas, C.; Pugh, B.F.; et al. ATF4 Gene Network Mediates Cellular Response to the Anticancer PAD Inhibitor YW3-56 in Triple-Negative Breast Cancer Cells. Mol. Cancer Ther. 2015, 14, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Clarke, R. Role of GRP78 in promoting therapeutic-resistant breast cancer. Future Med. Chem. 2015, 7, 1529–1534. [Google Scholar] [CrossRef]
- Rajapaksa, G.; Thomas, C.; Gustafsson, J.A. Estrogen signaling and unfolded protein response in breast cancer. J. Steroid. Biochem. Mol. Biol. 2016, 163, 45–50. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Welch, J.N.; Skaar, T.C. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol. Rev. 2001, 53, 25–71. [Google Scholar] [PubMed]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell Endocrinol. 2015, 418 Pt 3, 220–234. [Google Scholar] [CrossRef]
- Riggins, R.B.; Bouton, A.H.; Liu, M.C.; Clarke, R. Antiestrogens, aromatase inhibitors, and apoptosis in breast cancer. Vitam. Horm. 2005, 71, 201–237. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Sharma, C.G.; Jordan, V.C. Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells. Horm. Mol. Biol. Clin. Investig. 2010, 2, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Nichols, P.; Spicer, D.; Groshen, S.; Yu, M.C.; Lee, A.S. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006, 66, 7849–7853. [Google Scholar] [CrossRef]
- Wang, D.Y.; Fulthorpe, R.; Liss, S.N.; Edwards, E.A. Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol. Endocrinol. 2004, 18, 402–411. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef]
- Gomez, B.P.; Riggins, R.B.; Shajahan, A.N.; Klimach, U.; Wang, A.; Crawford, A.C.; Zhu, Y.; Zwart, A.; Wang, M.; Clarke, R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007, 21, 4013–4027. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tai, L.K.; Wong, L.L.; Putti, T.C.; Sethi, S.K.; Teh, M.; Koay, E.S. Proteomic characterization of differentially expressed proteins in breast cancer: Expression of hnRNP H1, RKIP and GRP78 is strongly associated with HER-2/neu status. Proteomics Clin. Appl. 2008, 2, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.T.; Rawale, S.; Allen, S.D.; Phillips, G.; Forni, G.; Morris, J.C.; Kaumaya, P.T. Novel engineered trastuzumab conformational epitopes demonstrate in vitro and in vivo antitumor properties against HER-2/neu. J. Immunol. 2007, 178, 7120–7131. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Cara, G.D.; Marengo, G.; Albanese, N.N.; Marabeti, M.R.; Musso, R.; Cancemi, P.; Pucci-Minafra, I. Proteomic profiling of Trastuzumab (Herceptin(R))-sensitive and -resistant SKBR-3 breast cancer cells. Anticancer Res. 2013, 33, 489–503. [Google Scholar]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef]
- Kumandan, S.; Mahadevan, N.R.; Chiu, K.; DeLaney, A.; Zanetti, M. Activation of the unfolded protein response bypasses trastuzumab-mediated inhibition of the PI-3K pathway. Cancer Lett. 2013, 329, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderwood, S.K. Heat shock proteins in breast cancer progression—A suitable case for treatment? Int J. Hyperthermia 2010, 26, 681–685. [Google Scholar] [CrossRef]
- Arrigo, A.P. Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones 2017, 22, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, M.R.; Matassa, D.S.; Sisinni, L.; Lettini, G.; Landriscina, M.; Esposito, F. TRAP1 revisited: Novel localizations and functions of a ‘next-generation’ biomarker (review). Int. J. Oncol. 2014, 45, 969–977. [Google Scholar] [CrossRef]
- Lettini, G.; Maddalena, F.; Sisinni, L.; Condelli, V.; Matassa, D.S.; Costi, M.P.; Simoni, D.; Esposito, F.; Landriscina, M. TRAP1: A viable therapeutic target for future cancer treatments? Expert. Opin. Ther. Targets 2017, 21, 805–815. [Google Scholar] [CrossRef]
- Matassa, D.S.; Amoroso, M.R.; Maddalena, F.; Landriscina, M.; Esposito, F. New insights into TRAP1 pathway. Am. J. Cancer Res. 2012, 2, 235–248. [Google Scholar] [PubMed]
- Amoroso, M.R.; Matassa, D.S.; Laudiero, G.; Egorova, A.V.; Polishchuk, R.S.; Maddalena, F.; Piscazzi, A.; Paladino, S.; Sarnataro, D.; Garbi, C.; et al. TRAP1 and the proteasome regulatory particle TBP7/Rpt3 interact in the endoplasmic reticulum and control cellular ubiquitination of specific mitochondrial proteins. Cell Death Differ. 2012, 19, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Amoroso, M.R.; Agliarulo, I.; Maddalena, F.; Sisinni, L.; Paladino, S.; Romano, S.; Romano, M.F.; Sagar, V.; Loreni, F.; et al. Translational control in the stress adaptive response of cancer cells: A novel role for the heat shock protein TRAP1. Cell Death Dis. 2013, 4, e851. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, F.; Sisinni, L.; Lettini, G.; Condelli, V.; Matassa, D.S.; Piscazzi, A.; Amoroso, M.R.; La Torre, G.; Esposito, F.; Landriscina, M. Resistance to paclitxel in breast carcinoma cells requires a quality control of mitochondrial antiapoptotic proteins by TRAP1. Mol. Oncol. 2013, 7, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisinni, L.; Maddalena, F.; Lettini, G.; Condelli, V.; Matassa, D.S.; Esposito, F.; Landriscina, M. TRAP1 role in endoplasmic reticulum stress protection favors resistance to anthracyclins in breast carcinoma cells. Int. J. Oncol. 2014, 44, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.D.; Yan, P.; Seidler, P.M.; Patel, H.J.; Sun, W.; Yang, C.; Que, N.S.; Taldone, T.; Finotti, P.; Stephani, R.A.; et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol. 2013, 9, 677–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, R.; Streller, F.; Scheel, A.H.; Ruschoff, J.; Reinert, M.C.; Dobbelstein, M.; Marchenko, N.D.; Moll, U.M. HER2/ErbB2 activates HSF1 and thereby controls HSP90 clients including MIF in HER2-overexpressing breast cancer. Cell Death Dis. 2014, 5, e980. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Kaneko, K.; Gwin, W.R.; Morse, M.A.; Hobeika, A.; Pogue, B.W.; Hartman, Z.C.; Hughes, P.F.; Haystead, T.; Lyerly, H.K. In Vivo Detection of HSP90 Identifies Breast Cancers with Aggressive Behavior. Clin Cancer Res. 2017, 23, 7531–7542. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chang, W.W.; Chen, Y.Y.; Tsai, Y.H.; Chou, Y.H.; Tseng, H.C.; Chen, H.L.; Wu, C.C.; Chang-Chien, J.; Lee, H.T.; et al. Hsp90alpha Mediates BMI1 Expression in Breast Cancer Stem/Progenitor Cells through Facilitating Nuclear Translocation of c-Myc and EZH2. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Stivarou, T.; Stellas, D.; Vartzi, G.; Thomaidou, D.; Patsavoudi, E. Targeting highly expressed extracellular HSP90 in breast cancer stem cells inhibits tumor growth in vitro and in vivo. Cancer Biol. Ther. 2016, 17, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Iwamoto, I.; Inaguma, Y.; Takizawa, T.; Nagata, K.; Asano, T.; Kato, K. Endoplasmic reticulum stress induces the phosphorylation of small heat shock protein, Hsp27. J. Cell Biochem. 2005, 95, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Kumano, M.; Furukawa, J.; Shiota, M.; Zardan, A.; Zhang, F.; Beraldi, E.; Wiedmann, R.M.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol. Cancer Ther. 2012, 11, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Livingston, J.A.; Wang, W.L.; Tsai, J.W.; Lazar, A.J.; Leung, C.H.; Lin, H.; Advani, S.; Daw, N.; Santiago-O’Farrill, J.; Hollomon, M.; et al. Analysis of HSP27 and the Autophagy Marker LC3B(+) Puncta Following Preoperative Chemotherapy Identifies High-Risk Osteosarcoma Patients. Mol. Cancer Ther. 2018, 17, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Qi, C.; Liu, B.; Lin, Y.; Fu, T.; Zeng, Q. Increased HSP27 correlates with malignant biological behavior of non-small cell lung cancer and predicts patient’s survival. Sci. Rep. 2017, 7, 13807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homaei-Shandiz, F.; Mehrad-Majd, H.; Tasbandi, M.; Aledavood, A.; Tavakol Afshari, J.; Ghavami, V.; Ghayour-Mobarhan, M. Anti-Heat Shock Protein-27 Antibody Levels in Women with Breast Cancer: Association with Disease Complications and Two-Year Disease-Free Survival. Asian Pac. J. Cancer Prev. 2016, 17, 4655–4659. [Google Scholar] [PubMed]
- Zhang, D.; Wong, L.L.; Koay, E.S. Phosphorylation of Ser78 of Hsp27 correlated with HER-2/neu status and lymph node positivity in breast cancer. Mol. Cancer 2007, 6, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.H.; Kang, K.W.; Kim, K.H.; Kwon, B.; Kim, S.K.; Lee, H.Y.; Kong, S.Y.; Lee, E.S.; Jang, S.G.; Yoo, B.C. Upregulated HSP27 in human breast cancer cells reduces Herceptin susceptibility by increasing Her2 protein stability. BMC Cancer 2008, 8, 286. [Google Scholar] [CrossRef]
- Diaz-Chavez, J.; Fonseca-Sanchez, M.A.; Arechaga-Ocampo, E.; Flores-Perez, A.; Palacios-Rodriguez, Y.; Dominguez-Gomez, G.; Marchat, L.A.; Fuentes-Mera, L.; Mendoza-Hernandez, G.; Gariglio, P.; et al. Proteomic profiling reveals that resveratrol inhibits HSP27 expression and sensitizes breast cancer cells to doxorubicin therapy. PLoS ONE 2013, 8, e64378. [Google Scholar] [CrossRef]
- Lee, C.H.; Hong, H.M.; Chang, Y.Y.; Chang, W.W. Inhibition of heat shock protein (Hsp) 27 potentiates the suppressive effect of Hsp90 inhibitors in targeting breast cancer stem-like cells. Biochimie 2012, 94, 1382–1389. [Google Scholar] [CrossRef]
- Miao, Y.R.; Eckhardt, B.L.; Cao, Y.; Pasqualini, R.; Argani, P.; Arap, W.; Ramsay, R.G.; Anderson, R.L. Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin. Cancer Res. 2013, 19, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Dobroff, A.S.; D’Angelo, S.; Eckhardt, B.L.; Ferrara, F.; Staquicini, D.I.; Cardo-Vila, M.; Staquicini, F.I.; Nunes, D.N.; Kim, K.; Driessen, W.H.P.; et al. Towards a transcriptome-based theranostic platform for unfavorable breast cancer phenotypes. Proc. Natl. Acad. Sci. USA 2016, 113, 12780–12785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Warri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisinni, L.; Pietrafesa, M.; Lepore, S.; Maddalena, F.; Condelli, V.; Esposito, F.; Landriscina, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance. Int. J. Mol. Sci. 2019, 20, 857. https://doi.org/10.3390/ijms20040857
Sisinni L, Pietrafesa M, Lepore S, Maddalena F, Condelli V, Esposito F, Landriscina M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance. International Journal of Molecular Sciences. 2019; 20(4):857. https://doi.org/10.3390/ijms20040857
Chicago/Turabian StyleSisinni, Lorenza, Michele Pietrafesa, Silvia Lepore, Francesca Maddalena, Valentina Condelli, Franca Esposito, and Matteo Landriscina. 2019. "Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance" International Journal of Molecular Sciences 20, no. 4: 857. https://doi.org/10.3390/ijms20040857
APA StyleSisinni, L., Pietrafesa, M., Lepore, S., Maddalena, F., Condelli, V., Esposito, F., & Landriscina, M. (2019). Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance. International Journal of Molecular Sciences, 20(4), 857. https://doi.org/10.3390/ijms20040857