New Insights into the Role of Epithelial–Mesenchymal Transition during Aging


Abstract
:
1. Introduction
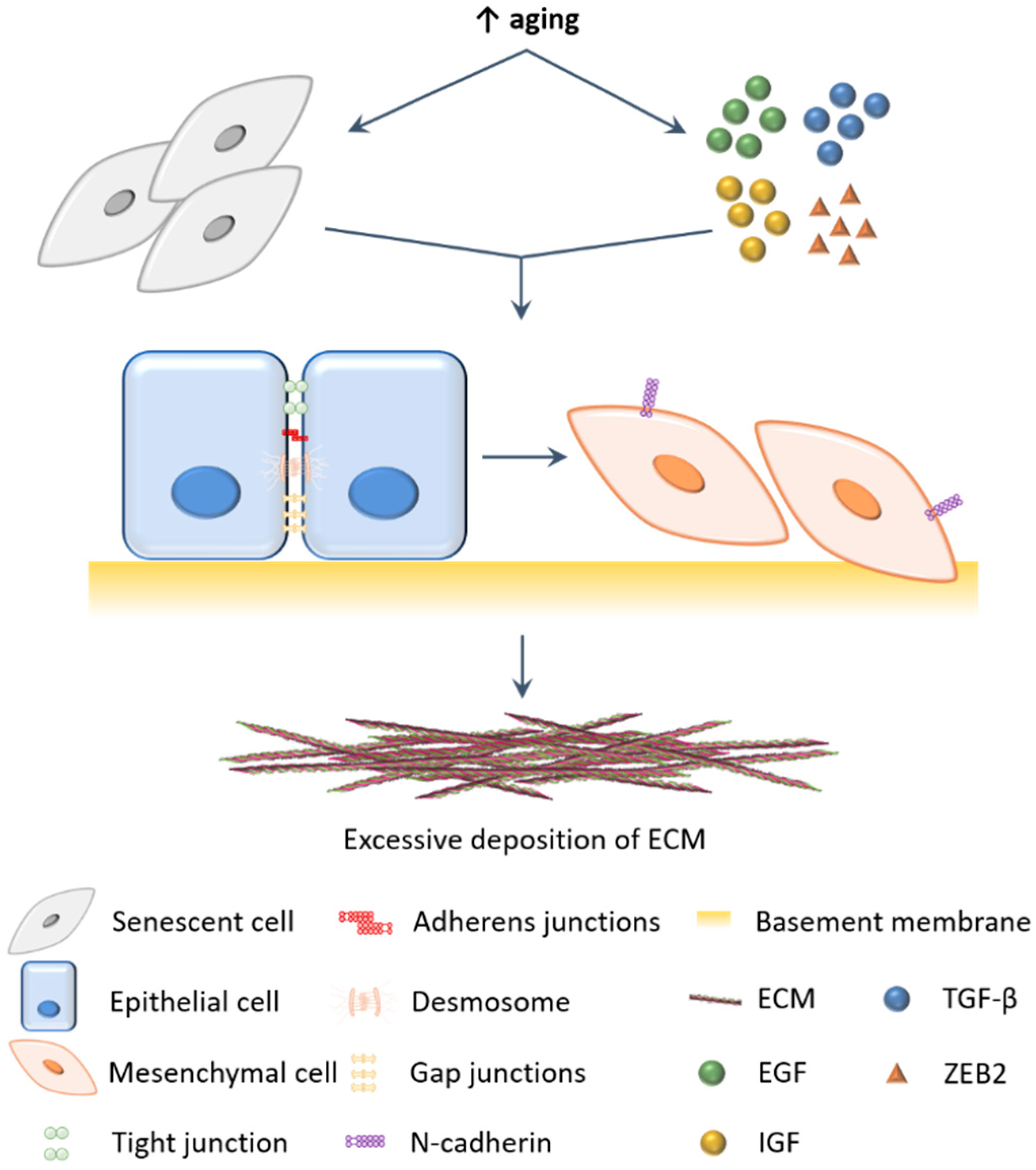
2. EMT Balance during Biological Aging
Is EMT Truly Involved in Liver Fibrosis?
3. EMT in Acquired Stem Cell Properties during Aging
4. Role of EMT during the Reprogramming of Aged Cells
5. Future Directions
Funding
Conflicts of Interest
References
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.Y.-J.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardes de Jesus, B.; Blasco, M.A. Telomerase at the intersection of cancer and aging. Trends Genet. 2013, 29, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Bernardes de Jesus, B.; Blasco, M.A. Aging by telomere loss can be reversed. Cell Stem Cell 2011, 8, 3–4. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef]
- Nieto, M.A. The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [Green Version]
- Bax, N.A.M.; Pijnappels, D.A.; van Oorschot, A.A.M.; Winter, E.M.; de Vries, A.A.F.; van Tuyn, J.; Braun, J.; Maas, S.; Schalij, M.J.; Atsma, D.E.; et al. Epithelial-to-mesenchymal transformation alters electrical conductivity of human epicardial cells. J. Cell. Mol. Med. 2011, 15, 2675–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 138, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Kishi, S.; Bayliss, P.E.; Hanai, J.-I. A prospective epigenetic paradigm between cellular senescence and epithelial-mesenchymal transition in organismal development and aging. Transl. Res. 2015, 165, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Craene, B.D.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, ZEB and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Spaderna, S.; Schmalhofer, O.; Wahlbuhl, M.; Dimmler, A.; Bauer, K.; Sultan, A.; Hlubek, F.; Jung, A.; Strand, D.; Eger, A.; et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008, 68, 537–544. [Google Scholar] [CrossRef]
- Whiteman, E.L.; Liu, C.J.; Fearon, E.R.; Margolis, B. The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene 2008, 27, 3875–3879. [Google Scholar] [CrossRef] [Green Version]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs Complex Couples Cell Density Sensing to Hippo-Dependent Control of the TGF-β-SMAD Pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef]
- Sousa-Franco, A.; Rebelo, K.; da Rocha, S.T.; Bernardes de Jesus, B. LncRNAs regulating stemness in aging. Aging Cell 2018, e12870. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, Y.; Muramatsu, T.; Uetake, H.; Kozaki, K.I.; Inazawa, J. miR-544a induces epithelial-mesenchymal transition through the activation of WNT signaling pathway in gastric cancer. Carcinogenesis 2015, 36, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jin, Z.Y.; Liu, C.H.; Xie, F.; Lin, X.S.; Huang, Q. MicroRNA-21 regulates biological behavior by inducing EMT in human cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 4684–4694. [Google Scholar]
- Bernardes de Jesus, B.; Marinho, S.P.; Barros, S.; Sousa-Franco, A.; Alves-Vale, C.; Carvalho, T.; Carmo-Fonseca, M. Silencing of the lncRNA Zeb2-NAT facilitates reprogramming of aged fibroblasts and safeguards stem cell pluripotency. Nat. Commun. 2018, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Vassilopoulos, A.; Fritz, K.S.; Petersen, D.R.; Gius, D. The human sirtuin family: Evolutionary divergences and functions. Hum Genom. 2011, 5, 485–496. [Google Scholar] [CrossRef]
- O’Callaghan, C.; Vassilopoulos, A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 2017, 16, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Correia, M.; Perestrelo, T.; Rodrigues, A.S.; Ribeiro, M.F.; Pereira, S.L.; Sousa, M.I.; Ramalho-Santos, J. Sirtuins in metabolism, stemness and differentiation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3444–3455. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Munoz-Martin, M.; Canamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, S.; Villanova, L.; Tanaka, S.; Aonuma, M.; Roy, N.; Berber, E.; Pollack, J.R.; Michishita-Kioi, E.; Chua, K.F. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 2015, 5, 9841. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ni, T.K.; Wronski, A.; Glass, B.; Skibinski, A.; Beck, A.; Kuperwasser, C. The SIRT2 Deacetylase Stabilizes Slug to Control Malignancy of Basal-like Breast Cancer. Cell Rep. 2016, 17, 1302–1317. [Google Scholar] [CrossRef] [Green Version]
- Miyo, M.; Yamamoto, H.; Konno, M.; Colvin, H.; Nishida, N.; Koseki, J.; Kawamoto, K.; Ogawa, H.; Hamabe, A.; Uemura, M.; et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br. J. Cancer 2015, 113, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.L.; Zhu, R.T.; Sun, Y.L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Danoff, T.M.; Kalluri, R.; Neilson, E.G. Early role of Fsp1 in epithelial-mesenchymal transformation. Am. J. Physiol. 1997, 273, F563–F574. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Jeon, H.S.; Song, E.K.; Han, M.K.; Park, S.I.; Lee, S.I.; Yun, H.J.; Kim, J.R.; Kim, J.S.; Lee, Y.C.; et al. CD40 ligation of rheumatoid synovial fibroblasts regulates RANKL-mediated osteoclastogenesis: Evidence of NF-kappaB-dependent, CD40-mediated bone destruction in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Li, Q.; Shi, G.; Li, N. Involvement of epithelial-mesenchymal transition in liver fibrosis. Saudi J. Gastroenterol. 2018, 24, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.; Brunet, A. Aging and reprogramming: A two-way street. Curr. Opin. Cell Biol. 2012, 24, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Frangogiannis, N.G.; Cardiology, D.; Einstein, A.; Ny, B. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-C.; Fogo, A.B. Fibrosis and renal aging. Kidney Int. Suppl. 2014, 4, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaaki, H.; Ichikawa, T.; Nakao, K.; Yatsuhashi, H.; Furukawa, R.; Ohba, K.; Omagari, K.; Kusumoto, Y.; Yanagi, K.; Inoue, O.; et al. Clinicopathological study of nonalcoholic fatty liver disease in Japan: The risk factors for fibrosis. Liver Int. 2008, 28, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Chen, S.Y.; Yeh, W.S.; Maroni, B.; Li, Q.; Lee, Y.C.; Collard, H.R. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001–2011. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Schmitt, R.; Cantley, L.G. The impact of aging on kidney repair. Am. J. Physiol. Ren. Physiol. 2008, 294, F1265–F1272. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cufí, S.; Vazquez-martin, A.; Oliveras-ferraros, C.; Joven, J.; Menendez, J.A. Metformin against TGF β-induced epithelial-to- mesenchymal transition (EMT): From cancer stem cells to aging-associated fibrosis. Cell Cycle 2010, 9, 4461–4468. [Google Scholar] [CrossRef] [PubMed]
- Neilson, E.G. Setting a trap for tissue fibrosis. Nat. Med. 2005, 11, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A Mesenchymal-to-Epithelial Transition Initiates and Is Required for the Nuclear Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. New Insights into Epithelial-Mesenchymal Transition in Kidney Fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Cai, G.-Y.; Ning, Y.-C.; Wang, J.-C.; Lv, Y.; Cui, S.-Y.; Fu, B.; Guo, Y.-N.; Chen, X.-M. Alleviation of senescence and epithelial-mesenchymal transition in aging kidney by short-term caloric restriction and caloric restriction mimetics via modulation of AMPK/mTOR signaling. Oncotarget 2017, 8, 16109–16121. [Google Scholar] [CrossRef] [Green Version]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson-Gilford progeria: Correlation with the vascular pathology of aging. Arter. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Aliper, A.M.; Csoka, A.B.; Buzdin, A.; Jetka, T.; Roumiantsev, S.; Moskalev, A.; Zhavoronkov, A. Signaling pathway activation drift during aging: Hutchinson-Gilford Progeria Syndrome fibroblasts are comparable to normal middle-age and old-age cells. Aging (Albany NY) 2015, 7, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/β-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Morali, O.G.; Delmas, V.; Moore, R.; Jeanney, C.; Thiery, J.P.; Larue, L. IGF-II induces rapid β-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene 2001, 20, 4942–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derada Troletti, C.; de Goede, P.; Kamermans, A.; de Vries, H.E. Molecular alterations of the blood-brain barrier under inflammatory conditions: The role of endothelial to mesenchymal transition. Biochim. Biophys. Acta 2016, 1862, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Podtelezhnikov, A.A.; Tanis, K.Q.; Nebozhyn, M.; Ray, W.J.; Stone, D.J.; Loboda, A.P. Molecular insights into the pathogenesis of Alzheimer’s disease and its relationship to normal aging. PloS ONE 2011, 6, e29610. [Google Scholar] [CrossRef]
- Hackett, T.L.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Pechkovsky, D.V.; Murray, L.A.; Argentieri, R.; Kicic, A.; Stick, S.M.; Bai, T.R.; et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am. J. Respir. Crit. Care Med. 2009, 180, 122–133. [Google Scholar] [CrossRef]
- Bernardes de Jesus, B.; Blasco, M.A. Assessing cell and organ senescence biomarkers. Circ. Res. 2012, 111, 97–109. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Awad, P.; Campisi, J.; Desprez, P.-Y. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Nakashiro, K.I.; Hayashi, Y.; Oyasu, R. Immunohistochemical expression of hepatocyte growth factor and c-Met/HGF receptor in benign and malignant human prostate tissue. Oncol. Rep. 2003, 10, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.; Naujokas, M.A.; Zuo, D.; Sangwan, V.; Frigault, M.M.; Petkiewicz, S.; Dankort, D.L.; Muller, W.J.; Park, M. HGF Converts ErbB2/Neu Epithelial Morphogenesis to Cell Invasion. Mol. Biol. Cell 2004, 16, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, S.; Wallenius, V.; Ekberg, S.; Brenzel, A.; Gressner, A.M.; Jansson, J.O. Insulin-like growth factors stimulate expression of hepatocyte growth factor but not transforming growth factor β1in cultured hepatic stellate cells. Endocrinology 1997, 138, 4683–4689. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Gohda, E.; Kaji, K.; Namba, M. Increased hepatocyte growth factor production by aging human fibroblasts mainly due to autocrine stimulation by interleukin-1. Biochem. Biophys. Res. Commun. 1998, 246, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A.J. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef] [Green Version]
- Hinkal, G.; Parikh, N.; Donehower, L.A. Timed somatic deletion of p53 in mice reveals age-associated differences in tumor progression. PLoS ONE 2009, 4, e6654. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Diehl, A.M. Epithelial-to-mesenchymal transitions in the liver. Hepatology 2009, 50, 2007–2013. [Google Scholar] [CrossRef] [Green Version]
- Taura, K.; Miura, K.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Penz-Osterreicher, M.; Brenner, D.A. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 2010, 51, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Kim, K.H.; Park, K.K. Mechanisms of fibrogenesis in liver cirrhosis: The molecular aspects of epithelial-mesenchymal transition. World J. Hepatol. 2014, 6, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Scholten, D.; Osterreicher, C.H.; Scholten, A.; Iwaisako, K.; Gu, G.; Brenner, D.A.; Kisseleva, T. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 2010, 139, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.; Kim, J.W.; Hui, J.J.; Li, Z.; Swain, G.P.; Fong, K.S.; Csiszar, K.; Russo, P.A.; Rand, E.B.; Furth, E.E.; et al. Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum. Pathol. 2008, 39, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Jung, Y.; Omenetti, A.; Abdelmalek, M.; Guy, C.D.; Yang, L.; Wang, J.; Witek, R.P.; Fearing, C.M.; Pereira, T.A.; et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 2009, 137, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Choi, S.S.; Bustamante, M.; McCall, S.J.; Perez, E.H.; Huang, J.; Li, Y.X.; Rojkind, M.; Diehl, A.M. Evidence for epithelial-mesenchymal transitions in adult liver cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G575–G583. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, S.; Hamzavi, J.; Ciuclan, L.; Godoy, P.; Ilkavets, I.; Ehnert, S.; Ueberham, E.; Gebhardt, R.; Kanzler, S.; Geier, A.; et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008, 135, 642–659. [Google Scholar] [CrossRef]
- Valdes, F.; Alvarez, A.M.; Locascio, A.; Vega, S.; Herrera, B.; Fernandez, M.; Benito, M.; Nieto, M.A.; Fabregat, I. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol. Cancer Res. 2002, 1, 68–78. [Google Scholar]
- Munker, S.; Wu, Y.L.; Ding, H.G.; Liebe, R.; Weng, H.L. Can a fibrotic liver afford epithelial-mesenchymal transition? World J. Gastroenterol. 2017, 23, 4661–4668. [Google Scholar] [CrossRef]
- Godoy, P.; Hengstler, J.G.; Ilkavets, I.; Meyer, C.; Bachmann, A.; Muller, A.; Tuschl, G.; Mueller, S.O.; Dooley, S. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology 2009, 49, 2031–2043. [Google Scholar] [CrossRef]
- Nitta, T.; Kim, J.S.; Mohuczy, D.; Behrns, K.E. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology 2008, 48, 909–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicchini, C.; Filippini, D.; Coen, S.; Marchetti, A.; Cavallari, C.; Laudadio, I.; Spagnoli, F.M.; Alonzi, T.; Tripodi, M. Snail controls differentiation of hepatocytes by repressing HNF4alpha expression. J. Cell Physiol. 2006, 209, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Robertson, H.; Marshall, H.L.; Pekalski, M.; Zhao, L.; Booth, T.A.; Jones, D.E.; Burt, A.D.; Kirby, J.A. Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Investig. 2008, 88, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Yoon, J. Schizandrin inhibits fibrosis and epithelial-mesenchymal transition in transforming growth factor-beta1-stimulated AML12 cells. Int. Immunopharmacol. 2015, 25, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.F.; Chang-Chien, P.W.; Chang, W.T.; Yeh, T.M.; Wang, J.Y. Propolis inhibits TGF-beta1-induced epithelial-mesenchymal transition in human alveolar epithelial cells via PPARgamma activation. Int. Immunopharmacol. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Inokuchi, S.; Roh, Y.S.; Song, J.; Loomba, R.; Park, E.J.; Seki, E. Transforming growth factor-beta signaling in hepatocytes promotes hepatic fibrosis and carcinogenesis in mice with hepatocyte-specific deletion of TAK1. Gastroenterology 2013, 144, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Pradere, J.P.; Affo, S.; Dapito, D.H.; Friedman, R.; Lefkovitch, J.H.; Schwabe, R.F. Epithelial Transforming Growth Factor-beta Signaling Does Not Contribute to Liver Fibrosis but Protects Mice From Cholangiocarcinoma. Gastroenterology 2016, 150, 720–733. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Omenetti, A.; Bass, L.M.; Anders, R.A.; Clemente, M.G.; Francis, H.; Guy, C.D.; McCall, S.; Choi, S.S.; Alpini, G.; Schwarz, K.B.; et al. Hedgehog activity, epithelial-mesenchymal transitions, and biliary dysmorphogenesis in biliary atresia. Hepatology 2011, 53, 1246–1258. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.S.; Omenetti, A.; Witek, R.P.; Moylan, C.A.; Syn, W.K.; Jung, Y.; Yang, L.; Sudan, D.L.; Sicklick, J.K.; Michelotti, G.A.; et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1093–G1106. [Google Scholar] [CrossRef] [Green Version]
- Omenetti, A.; Popov, Y.; Jung, Y.; Choi, S.S.; Witek, R.P.; Yang, L.; Brown, K.D.; Schuppan, D.; Diehl, A.M. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut 2008, 57, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Swiderska-Syn, M.; Syn, W.K.; Xie, G.; Kruger, L.; Machado, M.V.; Karaca, G.; Michelotti, G.A.; Choi, S.S.; Premont, R.T.; Diehl, A.M. Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy. Gut 2014, 63, 1333–1344. [Google Scholar] [CrossRef]
- Schultz, M.B.; Sinclair, D.A. When stem cells grow old: Phenotypes and mechanisms of stem cell aging. Development 2016, 143, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Moorefield, E.C.; Andres, S.F.; Blue, R.E.; Van Landeghem, L.; Mah, A.T.; Santoro, M.A.; Ding, S. Aging effects on intestinal homeostasis associated with expansion and dysfunction of intestinal epithelial stem cells. Aging (Albany NY) 2017, 9, 1898–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, R.M.; Blasco, M.A. Telomeres and telomerase in adult stem cells and pluripotent embryonic stem cells. Adv. Exp. Med. Biol. 2010, 695, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Munoz, P.; Flores, J.M.; Klatt, P.; Blasco, M.A. Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes Dev. 2007, 21, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Flores, I.; Benetti, R.; Blasco, M.A. Telomerase regulation and stem cell behaviour. Curr. Opin. Cell Biol. 2006, 18, 254–260. [Google Scholar] [CrossRef]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The longest telomeres: A general signature of adult stem cell compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, J.V.; Jones, K.M.; Basson, M.A.; Brack, A.S. The aged niche disrupts muscle stem cell quiescence. Nature 2012, 490, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Hsu, W.H.; Wang, C.C.; Chou, C.H.; Kuo, M.Y.; Lin, B.R.; Chen, S.T.; Tai, S.K.; Kuo, M.L.; Yang, M.H. Connective tissue growth factor activates pluripotency genes and mesenchymal-epithelial transition in head and neck cancer cells. Cancer Res. 2013, 73, 4147–4157. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1565. [Google Scholar] [CrossRef]
- Sato, R.; Semba, T.; Saya, H.; Arima, Y. Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets. Stem Cells 2016, 34, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Yamanaka, S. A fresh look at iPS cells. Cell 2009, 137, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.D.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Miyagoe-Suzuki, Y.; Yada, E.; Ito, N.; Nishiyama, T.; Nakamura, M.; Ono, Y.; Motohashi, N.; Segawa, M.; Masuda, S.; et al. Reprogramming efficiency and quality of induced Pluripotent stem cells (iPSCs) generated from muscle-derived fibroblasts of mdx mice at different ages. PLoS Curr. 2011, 3, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xie, J.; Shen, C.; Cheng, D.; Shi, Y.; Wu, Z.; Deng, X.; Chen, H.; Shen, B.; Peng, C.; et al. Upregulation of long noncoding RNA ZEB1-AS1 promotes tumor metastasis and predicts poor prognosis in hepatocellular carcinoma. Oncogene 2016, 35, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, H.; Qi, J.; Wang, L.; He, S.; Liu, J.; Feng, C.; Chen, C.; Li, W.; Guo, Y.; et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat. Cell Biol. 2013, 15, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Shen, L.; Yu, J.; Wan, H.; Guo, A.; Chen, J.; Long, Y.; Zhao, J.; Pei, G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 2011, 10, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, G.; Manabe, I.; Tsushima, K.; Fujiu, K.; Oishi, Y.; Imai, Y.; Maemura, K.; Miyagishi, M.; Higashi, Y.; Kondoh, H.; et al. DeltaEF1 mediates TGF-beta signaling in vascular smooth muscle cell differentiation. Dev. Cell 2006, 11, 93–104. [Google Scholar] [CrossRef]
- Grooteclaes, M.L.; Frisch, S.M. Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 2000, 19, 3823–3828. [Google Scholar] [CrossRef] [Green Version]
- Postigo, A.A.; Dean, D.C. ZEB, a vertebrate homolog of Drosophila Zfh-1, is a negative regulator of muscle differentiation. EMBO J. 1997, 16, 3935–3943. [Google Scholar] [CrossRef] [Green Version]
- Ponticos, M.; Partridge, T.; Black, C.M.; Abraham, D.J.; Bou-Gharios, G. Regulation of collagen type I in vascular smooth muscle cells by competition between Nkx2.5 and deltaEF1/ZEB1. Mol. Cell Biol. 2004, 24, 6151–6161. [Google Scholar] [CrossRef] [PubMed]
- Van Grunsven, L.A.; Taelman, V.; Michiels, C.; Opdecamp, K.; Huylebroeck, D.; Bellefroid, E.J. deltaEF1 and SIP1 are differentially expressed and have overlapping activities during Xenopus embryogenesis. Dev. Dyn. 2006, 235, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–29. [Google Scholar] [CrossRef] [PubMed]
- Angrand, P.O.; Vennin, C.; Le Bourhis, X.; Adriaenssens, E. The role of long non-coding RNAs in genome formatting and expression. Front. Genet. 2015, 6, 165. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-Driven mesenchymal-to-Epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Chen, J.; Gao, S.; Huang, J. LEIGC long non-coding RNA acts as a tumor suppressor in gastric carcinoma by inhibiting the epithelial-to-mesenchymal transition. BMC Cancer 2014, 14, 932. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Yu, Q.M.; Du, Y.A.; Yang, L.T.; Dong, R.Z.; Huang, L.; Yu, P.F.; Cheng, X.D. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int. J. Biol. Sci. 2013, 9, 587–597. [Google Scholar] [CrossRef]
- Huang, J.F.; Guo, Y.J.; Zhao, C.X.; Yuan, S.X.; Wang, Y.; Tang, G.N.; Zhou, W.P.; Sun, S.H. Hepatitis B virus X protein (HBx)-related long noncoding RNA (lncRNA) down-regulated expression by HBx (Dreh) inhibits hepatocellular carcinoma metastasis by targeting the intermediate filament protein vimentin. Hepatology 2013, 57, 1882–1892. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.H.; Lin, Y.S.; Chen, Y.; Yeh, C.T.; Huang, Y.L.; Hsieh, T.H.; Shieh, T.M.; Hsueh, C.; Chen, T.C. Long non-coding RNA AOC4P suppresses hepatocellular carcinoma metastasis by enhancing vimentin degradation and inhibiting epithelial-mesenchymal transition. Oncotarget 2015, 6, 23342–23357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Deng, F.; Qin, Y.; Zhao, Z.; Wu, Z.; Xing, Z.; Ji, A.; Wang, Q.J. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. 2016, 7, e2254. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Rongen, G.A.; Riksen, N.P. Metformin therapy in diabetes: The role of cardioprotection. Curr. Atheroscler. Rep. 2013, 15, 314. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Di Gennaro, E.; Bruzzese, F.; Avallone, A.; Budillon, A. New perspective for an old antidiabetic drug: Metformin as anticancer agent. Cancer Treat. Res. 2014, 159, 355–376. [Google Scholar] [CrossRef]
- Nasri, H.; Baradaran, A.; Ardalan, M.R.; Mardani, S.; Momeni, A.; Rafieian-Kopaei, M. Bright renoprotective properties of metformin: Beyond blood glucose regulatory effects. Iran J. Kidney Dis. 2013, 7, 423–428. [Google Scholar]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, J.; Xu, Z.; Feng, Y.; Zhang, M.; Liu, J.; Chen, R.; Shen, J.; Wu, J.; Lu, Z.; et al. Metformin is a novel suppressor for transforming growth factor (TGF)-beta1. Sci. Rep. 2016, 6, 28597. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.W.; Conrad, C.H. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J. Mol. Cell Cardiol. 2000, 32, 187–195. [Google Scholar] [CrossRef]
- Meyer, T.E.; Kovacs, S.J.; Ehsani, A.A.; Klein, S.; Holloszy, J.O.; Fontana, L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J. Am. Coll. Cardiol. 2006, 47, 398–402. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Organ | Characteristics | Reference |
|---|---|---|
 |
| [59] |
 |
| [45] |
 |
| [62,68] |
 |
| [45] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, F.; Moreira, C.; Nóbrega-Pereira, S.; Bernardes de Jesus, B. New Insights into the Role of Epithelial–Mesenchymal Transition during Aging. Int. J. Mol. Sci. 2019, 20, 891. https://doi.org/10.3390/ijms20040891
Santos F, Moreira C, Nóbrega-Pereira S, Bernardes de Jesus B. New Insights into the Role of Epithelial–Mesenchymal Transition during Aging. International Journal of Molecular Sciences. 2019; 20(4):891. https://doi.org/10.3390/ijms20040891
Chicago/Turabian StyleSantos, Francisco, Cristiana Moreira, Sandrina Nóbrega-Pereira, and Bruno Bernardes de Jesus. 2019. "New Insights into the Role of Epithelial–Mesenchymal Transition during Aging" International Journal of Molecular Sciences 20, no. 4: 891. https://doi.org/10.3390/ijms20040891
APA StyleSantos, F., Moreira, C., Nóbrega-Pereira, S., & Bernardes de Jesus, B. (2019). New Insights into the Role of Epithelial–Mesenchymal Transition during Aging. International Journal of Molecular Sciences, 20(4), 891. https://doi.org/10.3390/ijms20040891