Approaches towards Longevity: Reprogramming, Senolysis, and Improved Mitotic Competence as Anti-Aging Therapies


Abstract
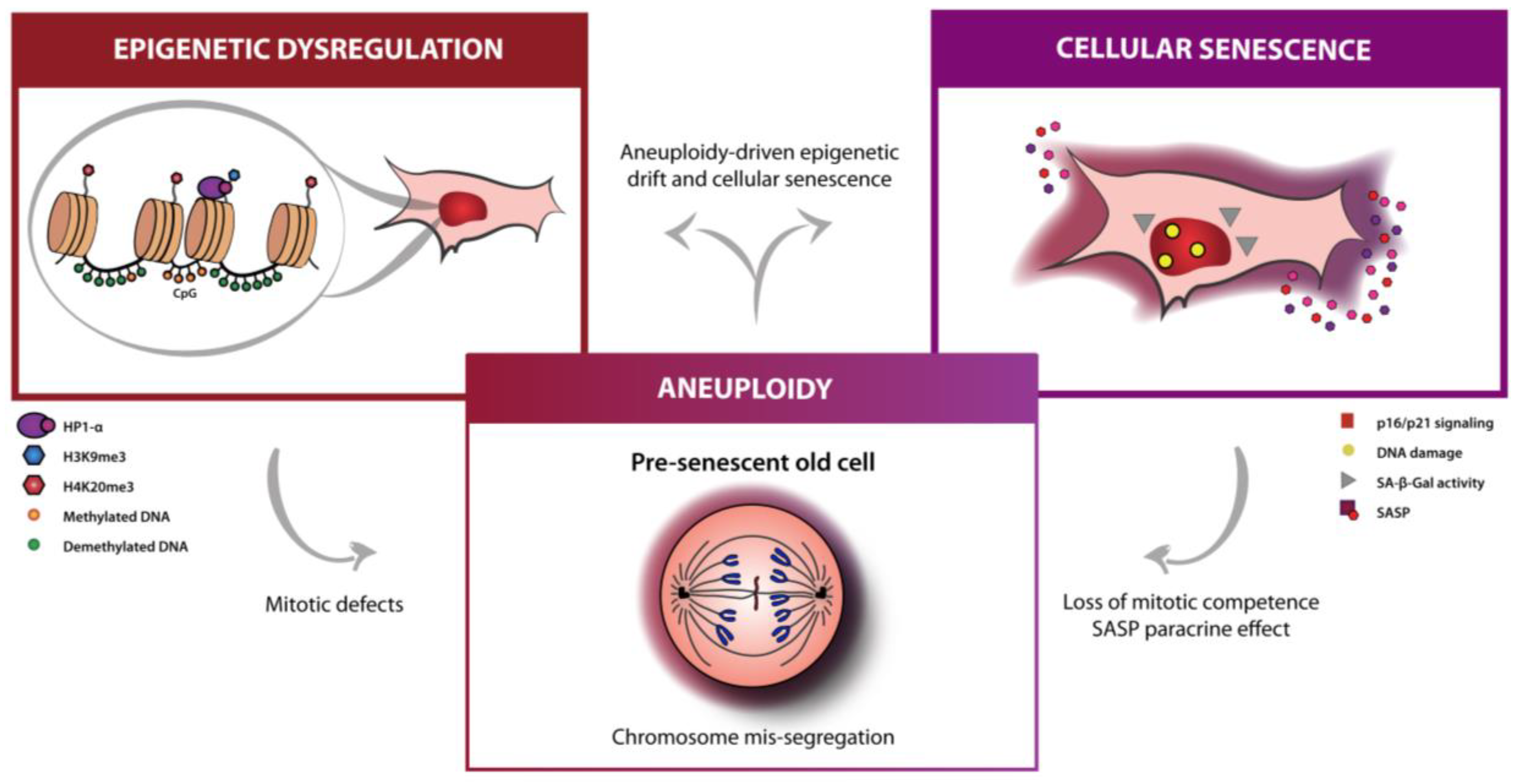
:1. Aging Hallmarks Explored in Anti-Aging Therapies: Epigenetics and Senescence
2. Aneuploidy as an Aging Hallmark Inter-Reliant with Senescence and Epigenetics
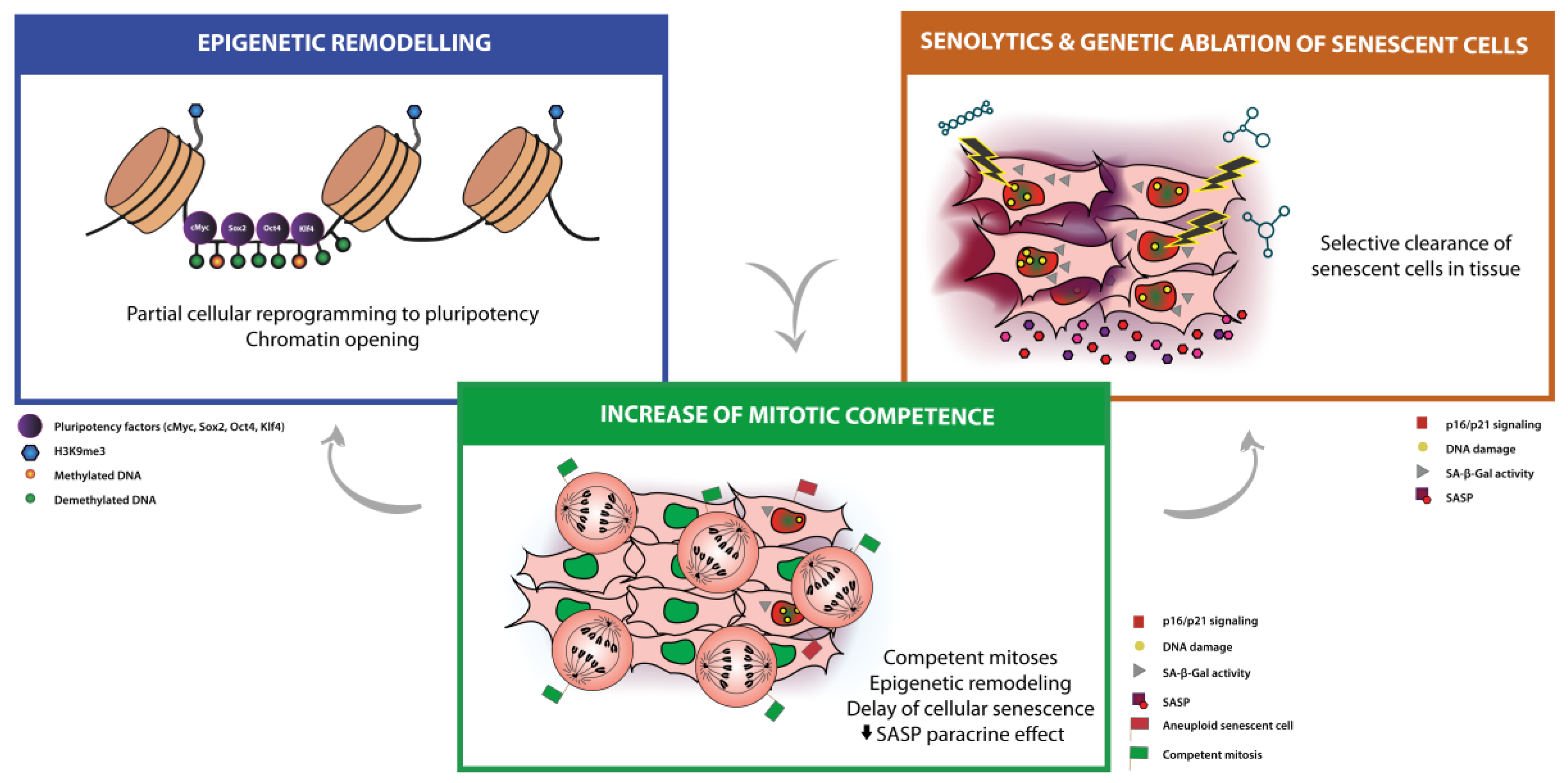
3. Mainstream Approaches for Anti-Aging Therapies: Partial Cellular Reprogramming and Senolysis
3.1. Partial Cellular Reprogramming
3.2. Senolytic Therapies and Clearance of Senescent Cells
4. Modulation of Mitotic Competence as a New Anti-Aging Therapy
5. Concluding Remarks and Future Directions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| mDNA | DNA methylation |
| HGPS | Hutchison-Gilford Progeria syndrome |
| H3K9me3 | H3 trimethylated on lysine 9 |
| H4K20me3 | H4 trimethylated on lysine 20 |
| HP1α | Heterochromatin protein 1α |
| DDR | DNA damage response |
| RB | Retinoblastoma |
| SA-β-Gal | Senescence-associated β-galactosidase |
| SASP | Senescence-associated secretory phenotype |
| GO | Gene ontology |
| dsDNA | Double-stranded DNA |
| cGAS-STING | Cyclic GMP-AMP synthase-stimulator of interferon genes |
| ALL | Acute lymphoblastic leukemia |
| iPSC | Induced pluripotent stem cells |
| XEN | Extra-embryonic endoderm |
| OSKM | Oct4, Sox2, Klf4, c-Myc |
| FOXO4 | Forkhead box protein O4 |
| hMSCs | Human mesenchymal stem cells |
| WS | Werner syndrome |
| ROS | Reactive oxygen species |
| CIN | Chromosomal instability |
| FoxM1 | Forkhead box M1 |
| HSCs | Hematopoietic stem cells |
| MuSCs | Muscle stem cells |
References
- Kirkwood, T.B.L. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Pollina, E.A.; Brunet, A. Epigenetic regulation of aging stem cells. Oncogene 2011, 30, 3105–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CPG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Teschendorff, A.E.; Maxwell, A.P.; Jacobs, I.J.; Kocjan, G.; Gayther, S.A.; Ramus, S.J.; Marth, C.; Beck, S.; Bell, C.G.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [Green Version]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.V.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Raj, K. The Epigenetic Clock and Aging. In Epigenetics of Aging and Longevity; Elsevier Inc.: Atlanta, GA, USA, 2018; pp. 95–118. [Google Scholar]
- Goldman, R.D.; Gruenbaum, Y.; Moir, R.D.; Shumaker, D.K.; Spann, T.P. Nuclear lamins: Building blocks. Genes Dev. 2002, 16, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Bhatia-Dey, N.; Kanherkar, R.R.; Stair, S.E.; Makarev, E.O.; Csoka, A.B. Cellular senescence as the causal nexus of aging. Front. Genet. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Shaulian, E.; Schreiber, M.; Piu, F.; Beeche, M.; Wagner, E.F.; Karin, M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell 2000, 103, 897–907. [Google Scholar] [CrossRef]
- Webley, K.; Bond, J.A.; Jones, C.J.; Blaydes, J.P.; Craig, A.; Hupp, T.; Wynford-Thomas, D. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol. Cell. Biol. 2000, 20, 2803–2808. [Google Scholar] [CrossRef]
- Qu, K.; Lin, T.; Wei, J.; Meng, F.; Wang, Z.; Huang, Z.; Wan, Y.; Song, S.; Liu, S.; Chang, H.; et al. Cisplatin induces cell cycle arrest and senescence via upregulating P53 and P21 expression in HepG2 cells. Nan Fang Yi Ke Da Xue Xue Bao 2013, 33, 1253–1259. [Google Scholar]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garré, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.I.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of macroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Kosar, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, E.; Pacifico, F.; Lavorgna, A.; De Palma, R.; D’Aiuto, E.; Palumbo, G.; Formisano, S.; Leonardi, A. NF-κB-dependent cytokine secretion controls Fas expression on chemotherapy-induced premature senescent tumor cells. Oncogene 2011, 30, 2707–2717. [Google Scholar] [CrossRef] [Green Version]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. XProgrammed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; VanSteeg, H.; Dollé, M.E.T.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, X.; Zhang, L. Negative regulation of inflammation by SIRT1. Pharmacol. Res. 2013, 67, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.; Weaver, R.; VanDeursen, J. P21 Both Attenuates and Drives Senescence and Aging in BubR1 Progeroid Mice. Cell Rep. 2013, 3, 1164–1174. [Google Scholar] [CrossRef]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.; Niederländer, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; Hiddinga, H.J.; et al. Opposing roles for p16Ink4aand p19Arfin senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Monti, D.; Ostan, R.; Borelli, V.; Castellani, G.; Franceschi, C. Inflammaging and human longevity in the omics era. Mech. Ageing Dev. 2017, 165, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C. Inflammaging as a major characteristic of old people: Can it be prevented or cured? Nutr. Rev. 2007, 65, S173–S176. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.A.; Marigorta, U.M.; Hughes, D.A.; Spataro, N.; Bosch, E.; Navarro, A. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat. Ecol. Evol. 2017, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, P.A.; Brunton, M.; Brown, W.M.C.; Doll, R.; Goldstein, H. Change of human chromosome count distributions with age: Evidence for a sex difference. Nature 1963, 197, 1080–1081. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.H.; McEwan, C.M. Total aneuploidy and age-related sex chromosome aneuploidy in cultured lymphocytes of normal men and women. Hum. Genet. 1977, 39, 329–337. [Google Scholar] [CrossRef]
- Guttenbach, M.; Schakowski, R.; Schmid, M. Aneuploidy and ageing: Sex chromosome exclusion into micronuclei. Hum. Genet. 1994, 94, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.B.; Alejandro, J.; Payne, S.; Thomas, S. Age-related aneuploidy analysis of human blood cells in vivo by fluorescence in situ hybridization (FISH). Mech. Ageing Dev. 1996, 90, 145–156. [Google Scholar] [CrossRef]
- Mukherjee, A.B.; Thomas, S. A Longitudinal Study of Human Age-Related Chromosomal Analysis in Skin Fibroblasts. Exp. Cell Res. 1997, 235, 8. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.F.; Sandberg, A.A. Sex chromosome aneuploidy and aging. Mutat. Res. DNAging 1995, 338, 107–113. [Google Scholar] [CrossRef]
- Macedo, J.C.; Vaz, S.; Logarinho, E. Mitotic dysfunction associated with aging hallmarks. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2017; Volume 1002, pp. 153–188. [Google Scholar]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Jeganathan, K.B.; Malureanu, L.; Perez-Terzic, C.; Terzic, A.; Van Deursen, J.M.A. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J. Cell Biol. 2006, 172, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Wijshake, T.; Malureanu, L.A.; Baker, D.J.; Jeganathan, K.B.; van de Sluis, B.; van Deursen, J.M. Reduced life- and healthspan in mice carrying a mono-allelic BubR1 MVA mutation. PLoS Genet. 2012, 8, e1003138. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Castillo, H.; Vasquez-Velasquez, A.I.; Rivera, H.; Barros-Nunez, P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: Delineation of clinical subtypes. Am. J. Med. Genet. A 2008, 146, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Macedo, J.C.; Vaz, S.; Bakker, B.; Ribeiro, R.; Bakker, P.L.; Escandell, J.M.; Ferreira, M.G.; Medema, R.; Foijer, F.; Logarinho, E. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 2018, 9, 2834. [Google Scholar] [CrossRef]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651.e5. [Google Scholar] [CrossRef] [PubMed]
- Andriani, G.A.; Almeida, V.P.; Faggioli, F.; Mauro, M.; Tsai, W.L.; Santambrogio, L.; Maslov, A.; Gadina, M.; Campisi, J.; Vijg, J.; et al. Whole Chromosome Instability induces senescence and promotes SASP. Sci. Rep. 2016, 6, 35218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. CGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, J.; Lilljebjorn, H.; Andersson, A.; Veerla, S.; Heldrup, J.; Behrendtz, M.; Fioretos, T.; Johansson, B. The DNA methylome of pediatric acute lymphoblastic leukemia. Hum. Mol. Genet. 2009, 18, 4054–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Xing, Z.; Yu, Y.E.; Tycko, B. Trans-acting epigenetic effects of chromosomal aneuploidies: Lessons from Down syndrome and mouse models. Epigenomics 2017, 9, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Mulla, W.A.; Seidel, C.W.; Zhu, J.; Tsai, H.J.; Smith, S.E.; Singh, P.; Bradford, W.D.; McCroskey, S.; Nelliat, A.R.; Conkright, J.; et al. Aneuploidy as a cause of impaired chromatin silencing and mating-type specification in budding yeast. Elife 2017, 6, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Murnane, J.P.; Zaki, B.I.; Compton, D.A. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014, 4, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef]
- Baker, D.J.; Dawlaty, M.M.; Wijshake, T.; Jeganathan, K.B.; Malureanu, L.; van Ree, J.H.; Crespo-Diaz, R.; Reyes, S.; Seaburg, L.; Shapiro, V.; et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat. Cell Biol. 2012, 15, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Hong, H.; Takahashi, K.; Yamanaka, S. Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat. Protoc. 2010, 5, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef]
- Kim, D.; Kim, C.H.; Moon, J. Il; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Li, X.; Liu, D.; Ma, Y.; Du, X.; Jing, J.; Wang, L.; Xie, B.; Sun, D.; Sun, S.; Jin, X.; et al. Direct Reprogramming of Fibroblasts via a Chemically Induced XEN-like State. Cell Stem Cell 2017, 21, 264–273.e7. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, L.; Mao, S.Q.; Li, Z.; Chen, J.; Zhang, R.R.; Wu, H.P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, K.; Zeng, X.; Yang, J.; Wu, Y.; Shi, X.; Qin, B.; Zeng, L.; Esteban, M.A.; Pan, G.; et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell 2011, 9, 575–587. [Google Scholar] [CrossRef]
- Chen, Z.; Chang, W.Y.; Etheridge, A.; Strickfaden, H.; Jin, Z.; Palidwor, G.; Cho, J.H.; Wang, K.; Kwon, S.Y.; Doré, C.; et al. Reprogramming progeria fibroblasts re-establishes a normal epigenetic landscape. Aging Cell 2017, 16, 870–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ät-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marión, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Karoubi, G.; Duchesneau, P.; Shutova, M.V.; Sung, H.K.; Tonge, P.; Bear, C.; Rogers, I.; Nagy, A.; Waddell, T.K. Generation of Induced Progenitor-like Cells from Mature Epithelial Cells Using Interrupted Reprogramming. Stem Cell Rep. 2017, 9, 1780–1795. [Google Scholar] [CrossRef] [PubMed]
- Göbel, C.; Goetzke, R.; Eggermann, T.; Wagner, W. Interrupted reprogramming into induced pluripotent stem cells does not rejuvenate human mesenchymal stromal cells. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Cañamero, M.; Rayon, T.; Ors, I.; Graña, O.; Megías, D.; Domínguez, O.; Martínez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 2018, e12877. [Google Scholar] [CrossRef]
- Tanabe, K.; Nakamura, M.; Narita, M.; Takahashi, K.; Yamanaka, S. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc. Natl. Acad. Sci. USA 2013, 110, 12172–12179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernig, M.; Lengner, C.J.; Hanna, J.; Lodato, M.A.; Steine, E.; Foreman, R.; Staerk, J.; Markoulaki, S.; Jaenisch, R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat. Biotechnol. 2008, 26, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, L.; Smith, J.R. Extension of the lifespan of cultured normal human diploid cells by vitamin E: A reevaluation. Proc. Natl. Acad. Sci. USA 1977, 74, 1640–1641. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S.I.; Clark, B.F. Kinetin delays the onset of ageing characteristics in human fibroblasts. Biochem. Biophys. Res. Commun. 1994, 201, 665–672. [Google Scholar] [CrossRef] [PubMed]
- McFarland, G.A.; Holliday, R. Retardation of the senescence of cultured human diploid fibroblasts by carnosine. Exp. Cell Res. 1994, 212, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, L.; Rattan, S.I.S.; Clark, B.F.C. Testing garlic for possible anti-ageing effects on long-term growth characteristics, morphology and macromolecular synthesis of human fibroblasts in culture. J. Ethnopharmacol. 1994, 43, 125–133. [Google Scholar] [CrossRef]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Roizen, M.F. Rapamycin fed late in life extends lifespan in genetically heterogenous mice. Yearb. Anesthesiol. Pain Manag. 2010, 460, 392–395. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Stroissnigg, F.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; James, L. New agents that target senescent cells the flavone, fisetin, and the BCL-X-L inhibitors. Aging 2017, 9, 955–963. [Google Scholar] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat. Rev. Drug Discov. 2018, 17, 377. [Google Scholar] [CrossRef]
- Sun, Y.; Coppé, J.P.; Lam, E.W.F. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, inflammation and immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Liu, Z.; Zhang, W.; Li, W.; Wu, Z.; Wang, W.; Ren, R.; Su, Y.; Wang, P.; Sun, L.; et al. Chemical screen identifies a geroprotective role of quercetin in premature aging. Protein Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, W.; Chang, L.; Han, Y.; Sun, L.; Gong, X.; Tang, H.; Liu, Z.; Deng, H.; Ye, Y.; et al. Vitamin C alleviates aging defects in a stem cell model for Werner syndrome. Protein Cell 2016, 7, 478–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, M.G.; Steinitz, A.; Geurts, J.; Pippenger, B.E.; Schaefer, D.J.; Martin, I.; Barbero, A.; Pelttari, K. Ascorbic acid attenuates senescence of human osteoarthritic osteoblasts. Int. J. Mol. Sci. 2017, 18, 2517. [Google Scholar] [CrossRef] [PubMed]
- Babu, J.R.; Jeganathan, K.B.; Baker, D.J.; Wu, X.; Kang-Decker, N.; Van Deursen, J.M. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 2003, 160, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Ricke, R.M.; Van Deursen, J.M. Aneuploidy in health, disease, and aging. J. Cell Biol. 2013, 201, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Ly, D.H.; Lockhart, D.J.; Lerner, R.A.; Schultz, P.G. Mitotic misregulation and human aging. Science 2000, 287, 2486–2492. [Google Scholar] [CrossRef]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Roose, J.; Clevers, H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997, 25, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hung, N.J.; Costa, R.H. Earlier expression of the transcription factor HFH-11B diminishes induction of p21CIP1/WAF1levels and accelerates mouse hepatocyte entry into S-phase following carbon tetrachloride liver injury. Hepatology 2001, 33, 1404–1414. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, V.V.; Lim, L.; Shin, B.; Costa, R.H.; Kalin, T. V; Meliton, L.; Meliton, A.Y.; Zhu, X.; Whitsett, J.A.; Kalinichenko, V.V.; et al. Differential expression of forkhead box transcription factors following butylated hydroxytoluene lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L695–L704. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Kelly, T.F.; Samadani, U.; Lim, L.; Rubio, S.; Overdier, D.G.; Roebuck, K.A.; Costa, R.H. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol. Cell. Biol. 1997, 17, 1626–1641. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-C.; Ustiyan, V.; Zhang, Y.; Cai, Y.; Kalin, T. V; Kalinichenko, V.V. Foxm1 transcription factor is required for the initiation of lung tumorigenesis by oncogenic KrasG12D. Oncogene 2014, 33, 5391–5396. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.C.; Zhang, Y.; Snyder, J.; Sutherland, M.J.; Burhans, M.S.; Shannon, J.M.; Park, H.J.; Whitsett, J.A.; Kalinichenko, V.V. Increased expression of FoxM1 transcription factor in respiratory epithelium inhibits lung sacculation and causes Clara cell hyperplasia. Dev. Biol. 2010, 347, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Balli, D.; Ustiyan, V.; Fulford, L.; Hiller, A.; Misetic, V.; Zhang, Y.; Paluch, A.M.; Waltz, S.E.; Kasper, S.; et al. Foxm1 Expression in Prostate Epithelial Cells Is Essential for Prostate Carcinogenesis. J. Biol. Chem. 2013, 288, 22527–22541. [Google Scholar] [CrossRef] [PubMed]
- Pfister, K.; Pipka, J.L.; Chiang, C.; Liu, Y.; Clark, R.A.; Keller, R.; Skoglund, P.; Guertin, M.J.; Hall, I.M.; Stukenberg, P.T. Identification of Drivers of Aneuploidy in Breast Tumors. Cell Rep. 2018, 23, 2758–2769. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Gogendeau, D.; Siudeja, K.; Gambarotto, D.; Pennetier, C.; Bardin, A.J.; Basto, R. Aneuploidy causes premature differentiation of neural and intestinal stem cells. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pfau, S.J.; Silberman, R.E.; Knouse, K.A.; Amon, A. Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes Dev. 2016, 30, 1395–1408. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 2008, 322, 703–709. [Google Scholar] [CrossRef]
- Patil, H.; Wilks, C.; Gonzalez, R.W.; Dhanireddy, S.; Conrad-Webb, H.; Bergel, M. Mitotic Activation of a Novel Histone Deacetylase 3-Linker Histone H1.3 Protein Complex by Protein Kinase CK2. J. Biol. Chem. 2016, 291, 3158–3172. [Google Scholar] [CrossRef] [PubMed]
- McManus, K.J.; Biron, V.L.; Heit, R.; Underhill, D.A.; Hendzel, M.J. Dynamic changes in histone H3 lysine 9 methylations: Identification of a mitosis-specific function for dynamic methylation in chromosome congression and segregation. J. Biol. Chem. 2006, 281, 8888–8897. [Google Scholar] [CrossRef]
- Herrera, L.A.; Prada, D.; Andonegui, M.A.; Dueñas-González, A. The epigenetic origin of aneuploidy. Curr. Genom. 2008, 9, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sun, P. The pathways to tumor suppression via route p38. Trends Biochem. Sci. 2007, 32, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kenny, A.E.; Rieder, C.L. P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol. Biol. Cell 2010, 21, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen-Macchi, A.; Sicora, O.; Kaczynska, K.; Oetken-Lindholm, C.; Pouwels, J.; Laine, L.; Kallio, M.J. Loss of p38gamma MAPK induces pleiotropic mitotic defects and massive cell death. J. Cell Sci. 2011, 124, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.; Shinohara, M.; Rieder, C.L. The p38-Mediated Stress-Activated Checkpoint: A Rapid Response System for Delaying Progression through Antephase and Entry into Mitosis. Cell Cycle 2005, 4, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.; Wang, X.; Li, H.; Fornace, A.J. A functional role for p38 MAPK in modulating mitotic transit in the absence of stress. J. Biol. Chem. 2007, 282, 22984–22992. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Study | Therapeutic/Preventive Rejuvenation Strategy | Epigenetic Modulation | Decrease in Cellular Senescence | SASP Modulation | Improvement of Cell Proliferative Fitness | Ref. |
|---|---|---|---|---|---|---|
| Reprogramming | ||||||
| Esteban 2010 | Vitamin C promoted generation of mouse and human iPSCs | √ | √ | [91] | ||
| Wang 2011 | Histone demethylases Jhdm1a/1b identified as key effectors in vitamin C induced reprogramming | √ | √ | √ | [92] | |
| Liu 2011 | Reprogramming of HGPS cells alleviated progeroid phenotypes | √ | √ | √ | [94] | |
| Ocampo 2016 | Transient expression of OSKM factors alleviated age-associated symptoms, prolonged lifespan in progeroid mice and improved tissue homeostasis in older mice | √ | √ | √ | √ | [97] |
| Senolysis | ||||||
| Baker 2011 | Long-life and late-life ablation of p16-positive cells delayed or attenuated progression of age-related disorders | √ 2 | √ | √ | [48] | |
| Jeon 2017 | Ablation of p16-positive cells/ use of senolytic compound UBX0101 attenuated the development of post-traumatic osteoarthritis and created a pro-regenerative environment | √ 2 | √ | √ | [143] | |
| Xu 2018 | Combination of Quercetin + Dasatinib extended both health- and lifespan in aged mice | √ | √ | √ 1 | [122] | |
| Geng 2018 | Quercetin rejuvenated WS, HGPS and chronologically-aged hMSCs | √ | √ | √ | √ | [127] |
| Li 2016 | Vitamin C rejuvenated WS hMSCs | √ | √ | √ | √ | [128] |
| Burger 2017 | Vitamin C attenuated senescence of human osteoarthritic osteoblasts | √ | √ | [129] | ||
| Chang 2016 | ABT263-induced senescent cell clearance and rejuvenated aged hematopoietic stem cells (HSCs) and muscle stem cells (MuSCs) | √ 2 | √ | √ | [116] | |
| Fuhrmann-Stroissnigg 2017 | HSP90 inhibitor 17-DMAG delayed onset of age-associated symptoms in a progeroid mouse model | √ 2 | √ | √ | [118] | |
| Mitotic Competence | ||||||
| Baker 2012 | High-level expression of BubR1 extended lifespan and delayed age-related deterioration and aneuploidy in several tissues | √ | √ | [83] | ||
| Macedo 2018 | Restoring levels of FoxM1 in elderly and HGPS cells reestablished mitotic proficiency and reduced senescence | √ | √ | √ | √ | [66] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melo Pereira, S.; Ribeiro, R.; Logarinho, E. Approaches towards Longevity: Reprogramming, Senolysis, and Improved Mitotic Competence as Anti-Aging Therapies. Int. J. Mol. Sci. 2019, 20, 938. https://doi.org/10.3390/ijms20040938
Melo Pereira S, Ribeiro R, Logarinho E. Approaches towards Longevity: Reprogramming, Senolysis, and Improved Mitotic Competence as Anti-Aging Therapies. International Journal of Molecular Sciences. 2019; 20(4):938. https://doi.org/10.3390/ijms20040938
Chicago/Turabian StyleMelo Pereira, Sofia, Rui Ribeiro, and Elsa Logarinho. 2019. "Approaches towards Longevity: Reprogramming, Senolysis, and Improved Mitotic Competence as Anti-Aging Therapies" International Journal of Molecular Sciences 20, no. 4: 938. https://doi.org/10.3390/ijms20040938
APA StyleMelo Pereira, S., Ribeiro, R., & Logarinho, E. (2019). Approaches towards Longevity: Reprogramming, Senolysis, and Improved Mitotic Competence as Anti-Aging Therapies. International Journal of Molecular Sciences, 20(4), 938. https://doi.org/10.3390/ijms20040938