1. Introduction
The skin is an organ specialized as a first major barrier against ionizing radiations and chemical damage. Its complete exposure to environmental DNA damaging agents renders it susceptible of developing different pathologies, including cancer. Skin tumours are the most common form of cancer, and its deadliest type—melanoma—exhibits an increasing incidence and accounts for ~1% of all cancer-related deaths worldwide (World Health Organization Annual Report, 2017).
Ultraviolet ionizing radiation (UVR) constitutes ~10% of total sunlight output, and is the most prominent causative factor for skin cancers (reviewed in open online resources: In.
Ultraviolet Waves.; 2010). Its long- and medium-wave components A and B (UVA, wavelength 320–400 nm and UVB, wavelength 290–320 nm)—those most penetrant through the ozone layer—can induce DNA damage directly, by promoting the formation of specific DNA products (e.g., pyrimidine-pyrimidine dimers, most often formed by the crosslinking of thymine pairs). Moreover, telomeres are particularly sensitive to UVR damage due to their relative enrichment in target TT and G bases, amplifying the genome instability resulting from genome-wide point alterations [
1]. In addition, UVR can damage DNA and other cell structures through the accumulation of reactive oxygen species (ROS), which in turn favour further damage upon activation of inflammatory signalling and disruption of mitochondrial function. Surviving unrepaired damaged cells can initiate tumours or contribute to skin aging (reviewed in [
2,
3]).
Of note, a number of chemicals can non-specifically enhance the toxicity of skin cells by rising basal metabolic/oxidative stress. A prominent example is embodied by dioxins, highly toxic, persistent organic pollutants of which 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a prototypical species. Dioxins exert their toxicity by acting as potent ligands of the bHLH transcriptional regulator aryl hydrocarbon receptor (AhR), a pivotal xenobiotic sensor in eukaryotic cells that, upon activation, can alter global gene expression patterns, and trigger metabolic programmes that amplify the accumulation of toxic compounds [
4].
Given current habits favouring outdoor activities and the pervasiveness of pollutants with phototoxic potential, identifying natural compounds with potential protective activity is warranted. The Antarctic hair grass
Deschampsia antarctica is a tracheophyte capable of thriving under extreme weather conditions, including high oxygen tension and solar radiation [
5]. One of only two flowering plants in Antarctica, it partly owes its resilience to secondary metabolism routes, which provide photoquenching compounds as well as phenolic substances with strong antioxidant potential, including flavonoids such as apigenin and luteolin [
6]. Previous studies on soluble extracts of
Deschampsia antarctica (hereon EDA) support that these activities can be transferred as antioxidant and antiaging properties on human cells [
7]. Therefore, these preparations have the potential to be used as protective supplements against environmental aggressions, but the characterization of their specific activity in the face of specific agents is still lacking.
Matrix metalloproteinases (MMPs) constitute a heterogeneous family of enzymes capable of hydrolyzing collagen and degrading different components of the ECM, and are involved in several physiological and pathological processes. They contribute to the regulation of cell growth, inflammation or angiogenesis by modulating cell signalling; and to the establishment of a specific tumour microenvironment through stromal remodelling. Their activity is tightly regulated by endogenous inhibitors the tissue inhibitor of metalloproteinases (TIMPs). The activity of MMPs has been specifically associated with photoageing [
8]. A prominent member is the collagenase MMP1, an ubiquitous, potent MMP capable of degrading collagens I, II and III that is upregulated by different sources of cell stress [
9].
Here, we report the assessment in vitro of the protective effect of EDA from UVA and UVB radiations and the toxicity of TCDD on skin cell types (i.e., skin fibroblasts and keratinocytes). Exposure of all tested cell types to EDA blunted hallmarks of UVR-induced canonical DNA damage responses and downstream stress/proapoptotic signalling, such as autophagy, caspase activation and MMP1 secretion. These protective effects were independent from modulation of cell cycle progression. Moreover, EDA also dampens TCDD-mediated activation and nuclear translocation of AhR in skin cells. Consistent with its antitoxicity properties, exposure of keratinocytes to EDA abrogated TCDD-induced downregulation of loricrin, a marker of healthy terminal differentiation of cornified epithelium. Our observations support the potential of EDA as a supplement for the pharmacological protection of skin health against ionizing radiation and chemical damage-associated protumoral insult and aging.
3. Discussion
Phototoxicity from ultraviolet ionizing radiations is a permanent environmental aggression to skin, and a direct cause of skin aging and tumorigenesis. Different factors derived from modern lifestyles (increased age expectancy; promotion of outdoor activity and travelling to different latitudes; increasing presence of environmental contaminants with toxic potential) challenge skin health and require the development of technologies that minimize their impact. Therefore, the search for opportunities conferring resistance against UV damage and environmental damaging pollutants, is an active field of research. Natural extracts and compounds are particularly interesting, because they have typically evolved low toxicity and high anti-stressor activities, and very often combine multiple synergistic effects such as antioxidant and enhancer of tissue repair. In this study, we report the capacity of a hydrophilic extract from
Deschampsia antarctica (EDA) to reduce toxicity induced by UV light on skin fibroblasts and keratinocytes as inferred from different molecular markers, including DNA damage-associated γH2AX, PARP cleavage, induction of autophagy and secretion of MMP1. Of note, EDA was also capable of blunting the toxicity of TCDD, a potent dioxin that is known to amplify the damage and subsequent carcinogenic effect of other agents [
16].
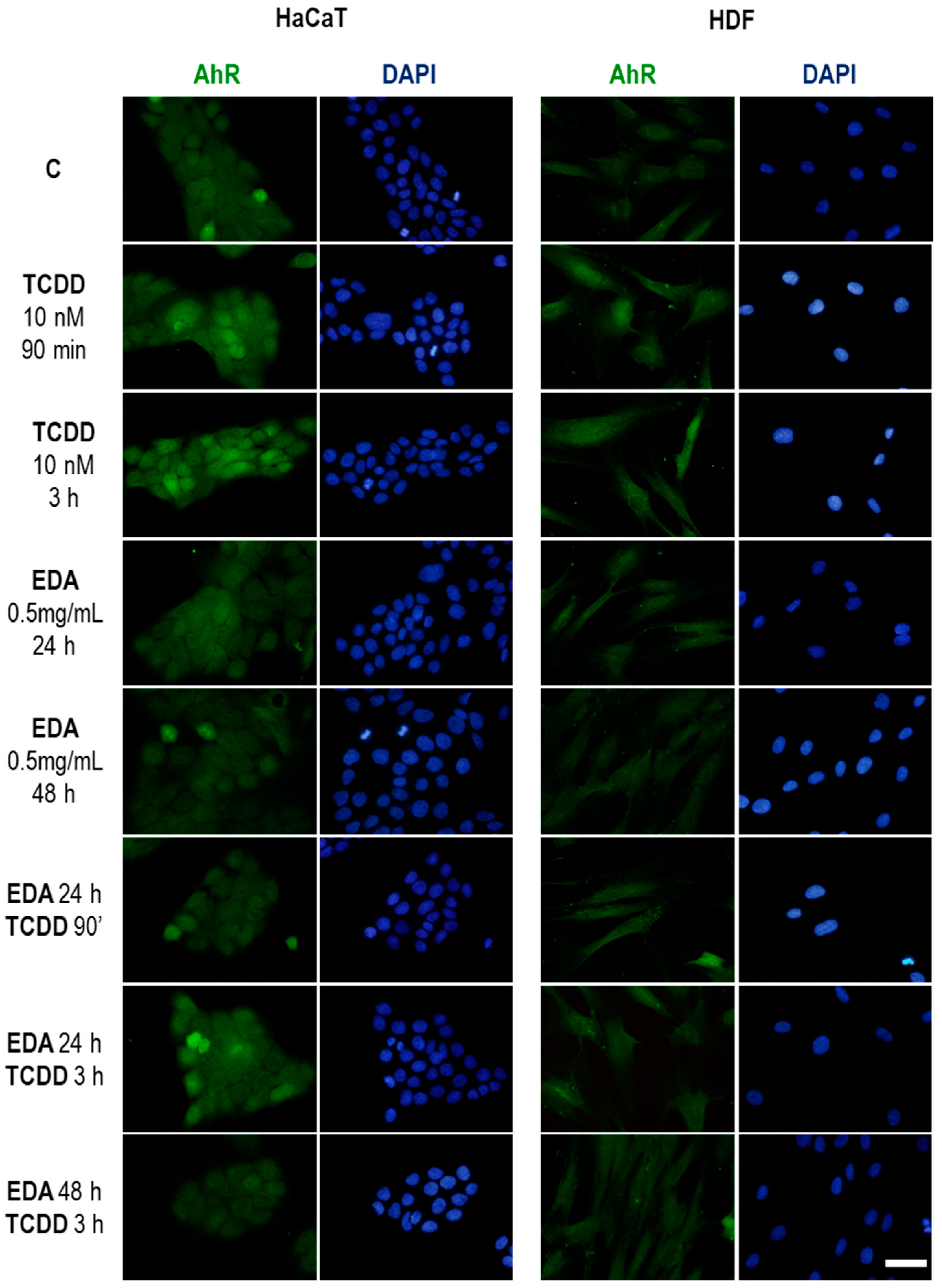
The inhibition of TCDD toxicity by EDA was not only evaluated through indirect proxies for skin cell homeostasis (proliferation, morphology, loricrin expression) but also directly by assessing the expression levels and translocation of the TCDD receptor in the cell, AhR. While EDA could boost degradation mechanisms targeting TCDD independently of AhR, it is tempting to speculate that EDA exerts direct regulation on AhR, possibly through proteostatic mechanisms such as Hsp90, given its anti-inflammatory activity in vitro [
21]. Of note, these mechanisms might also contribute to the protective effect of EDA against phototoxicity.
Generic mechanisms such as antioxidant activity from phenolic compounds in EDA could partly explain a blockade of DNA damage and downstream responses [
22], because dysregulated ROS accumulation is both a hallmark of phototoxicity as well as a major cause for the oxidative damage of genomic material and other structures such as lipids. Nonetheless, a non-exclusive protective mechanism likely at play is the direct quenching of ionizing radiation, because apart from adaptations of its photosynthetic machinery,
Deschampsia antarctica plants produce substantial amounts of carotenoids such as zeaxanthin and other xanthophyll cycle intermediates [
23], which are known to confer photoprotection. Future studies may characterize isolate contributions from different components in EDA to specific mechanisms of cell protection.
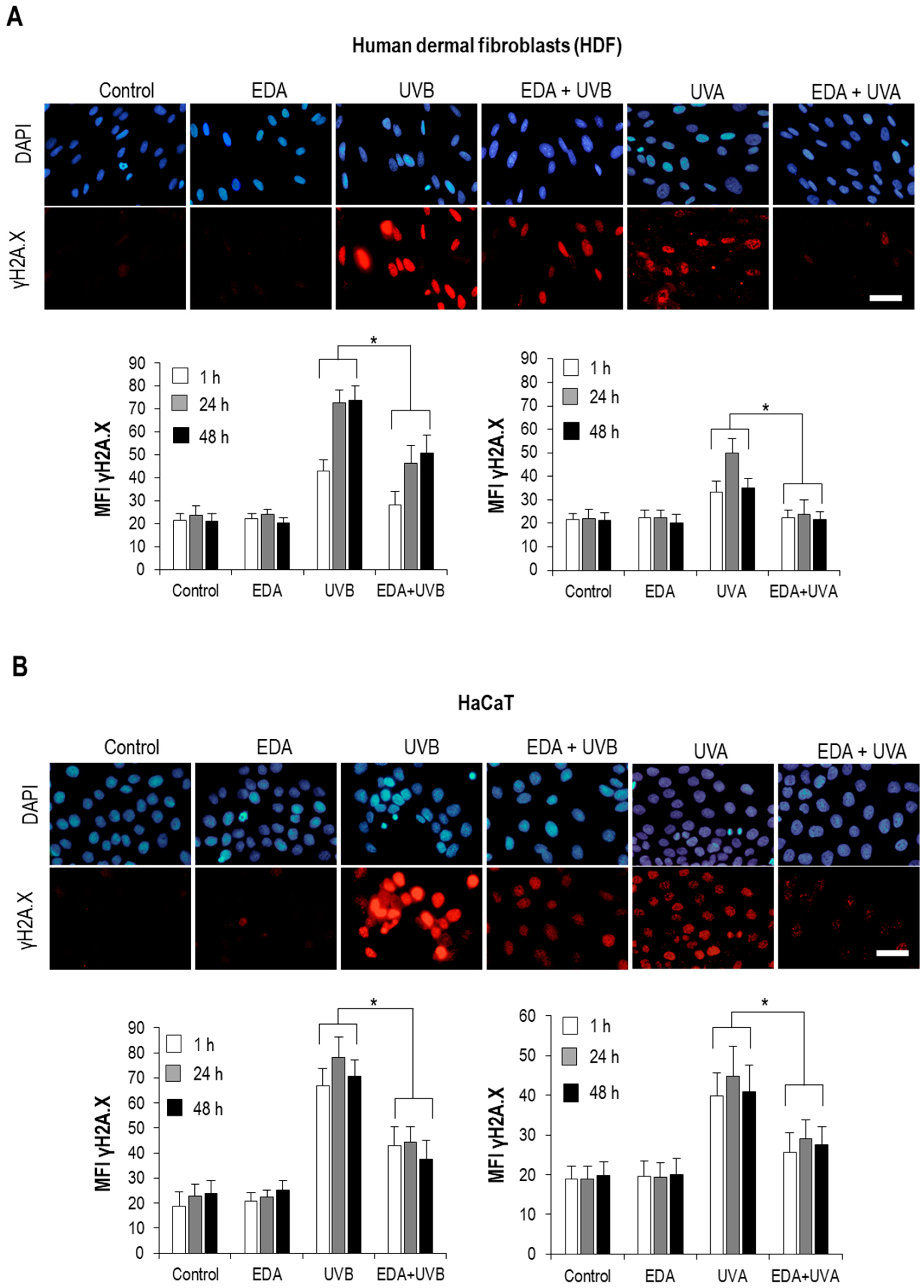
Interestingly, our observations support that while UV-induced apparent DNA damage is significantly diminished by simultaneous exposure to EDA, checkpoint mechanisms are still active and cells undergo blockade of their progression through the cell cycle when exposed to UVB light regardless of the presence or absence of EDA. Specifically, analysis of unsynchronized cell cultures revealed that UVB light elicited an accumulation of cells in S and G2/M phases, and simultaneous exposure to EDA did not significantly diminish this effect. Although the precise mechanisms at play will require extensive mechanistic characterization, beyond the scope of this report, it is clear that certain mechanisms sensing and transducing the impact of UVB on the cell are active despite the presence of EDA. While our assays monitoring stress signalling such as PARP cleavage and autophagy clearly show a protective effect from EDA, a possibility exists that surges in ROS are only partially lowered to levels significantly less harmful, but nonetheless detectable by specific mechanisms such as sestrins [
24]. Another non-exclusive explanation would lie on the hypothesis that certain molecules in the cell distinct from DNA are still damaged despite the presence of EDA; a good candidate would be membrane lipids, which face UV radiation at peripheral regions of the cell [
25]. Beyond the specific mechanisms at play, our observations in this regard bear relevance because they highlight that checkpoint mechanisms are still effective in cells undergoing stress protection by EDA, a feature of particular importance when considering the antitumoral effect from these preparations.
In summary, we contribute evidence that EDA displays significant protective roles in both fibroblasts and keratinocytes in the face of both UV radiation and prototypical dioxin toxicity, while having little intrinsic impact on cell proliferation and homeostasis. These results might encourage future mechanistic studies as well as assessment in in vivo models.
4. Materials and Methods
4.1. Reagents
Deschampsia antarctica extract was obtained from Cantabria Labs, Madrid, Spain. The methods for obtaining the extract have been previously published [
7]. Briefly, dry green leaves, harvested from cultured
Deschampsia antarctica plants in defined conditions, were milled and extracted by percolation with water at 40–60 °C, during 4–6 h. The extract was filtered through a 1μm filter and lyophilized. This stock was prepared in the culture medium to the desired concentration.
Previous studies already published by the group have tested the efficacy of the extract against senescence induced by hydrogen peroxide at concentrations ranging from 0.3 through to 1 mg/mL [
7]. Based on these studies, we treated fibroblasts and keratinocytes under the following conditions: 0.5 mg/mL of EDA for 24 h before exposition to environmental aggressive agents. For detection of MMP1, HDF were treated with 0.1 and 0.3 mg/mL EDA for 24 h.
4.2. Cell Cultures
Human dermal fibroblasts (HDF) were obtained from a skin biopsy after two rounds of trypsinization with 0.25% Trypsin-EDTA by shaking at 37° C. We also used the keratinocyte cell line HaCaT. Cells were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and penicillin (50 μg/mL) and streptomycin (50 μg/mL) (HyClone Laboratories, South Logan, UT, USA). Cells were maintained at 37 °C, 95% humidity and 5% CO2 in an incubator (Heraeus HERAcell, Thermo Scientific, Waltham, MA, USA).
4.3. Irradiation
HDF and HaCaT were subjected to UVA and UVB radiation. The sources of UVA and UVB lights were a lamp of 300–400 nm and 15.9 W/m
2 (CAMAG) and a lamp of 270–380 nm, which was filtrated by glass filter of 305 ± 5 nm (Newport, Irvine, CA, USA) respectively. The spectra of the UVA and UVB lamps were recorded and measured using a radiometer USB2000+ (ocean Optics, Dumedin, FL, USA) (
Figure S3). UV dose-response curves were carried out (
Figure S2) and different doses were used in most of the assays: 700 mJ/cm
2 and 300 mJ/cm
2 in the case of UVB for HDF and HaCaT cells respectively, and 3000 mJ/cm
2 in the case of UVA for both cell types. For MMP1 detection assays HDF cells were irradiated with UVB light at lower dose: 20 and 40 mJ/cm
2, to induce detectable cell stress while avoiding the triggering of proapoptotic signalling.
For irradiation, cell culture medium was replaced by PBS in all cases. Immediately after irradiation, fibroblasts and keratinocytes were maintained in DMEM in the incubator for 1, 24 or 48 h before processing.
4.4. Dioxin: Tetrachlorodibenzo-p-dioxin (TCDD)
Different concentrations of TCDD (15–300 nM) were tested on human dermal fibroblasts and keratinocytes at different incubation times, always without exceeding a final DMSO concentration of 0.1% (Sigma, Saint Louis, MO, USA).
4.5. Cell Morphology and Viability Analysis
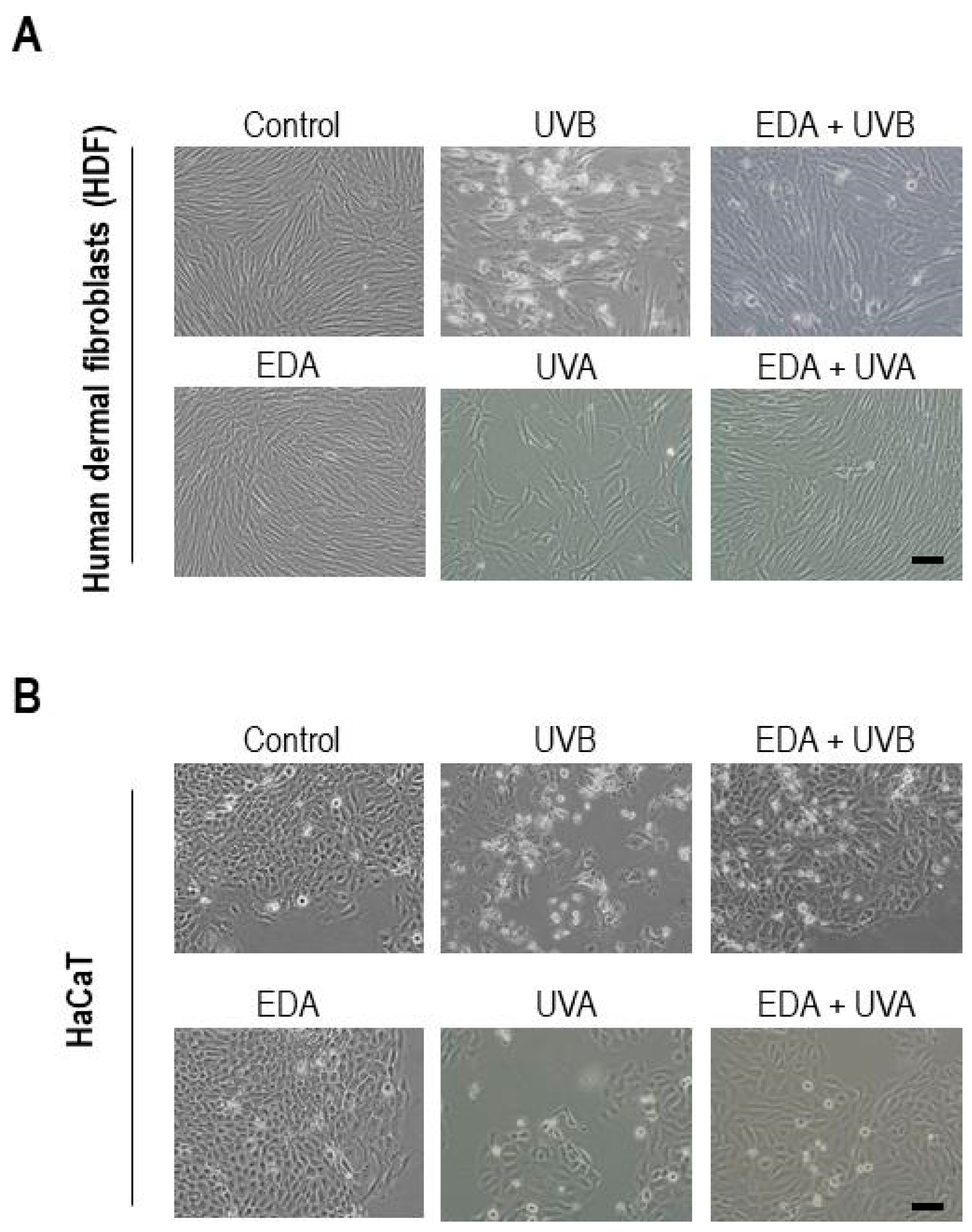
Cell cultures were observed at different time points (0, 24 and 48 h) after EDA, UV/TCDD and EDA+UV/TCDD treatments using an inverted microscope (Olympus IX51, Olympus Surgical Technologies Europe, Hamburg, Germany). Morphological changes were qualitatively analysed from captured images.
Cell viability in HaCaT and HDF cells after different treatments was determined by MTT assay. For that, cells 24 h after the treatments, were incubated with MTT (50 μg/mL) for 3 h. The formazan crystals were dissolved with DMSO and the optical density was determined in a SpectraFluor plate reader (Tecan Trading AG, Switzerland) at a wavelength of 542 nm.
Cell proliferation in HaCaT and HDF cells after different treatments was measured by crystal violet staining (0.2% (w/v) in 2% Ethanol) for 20 min. Optical density was determined in a SpectraFluor plate reader (Tecan) at a wavelength of 570 nm.
4.6. Immunofluorescence Assays
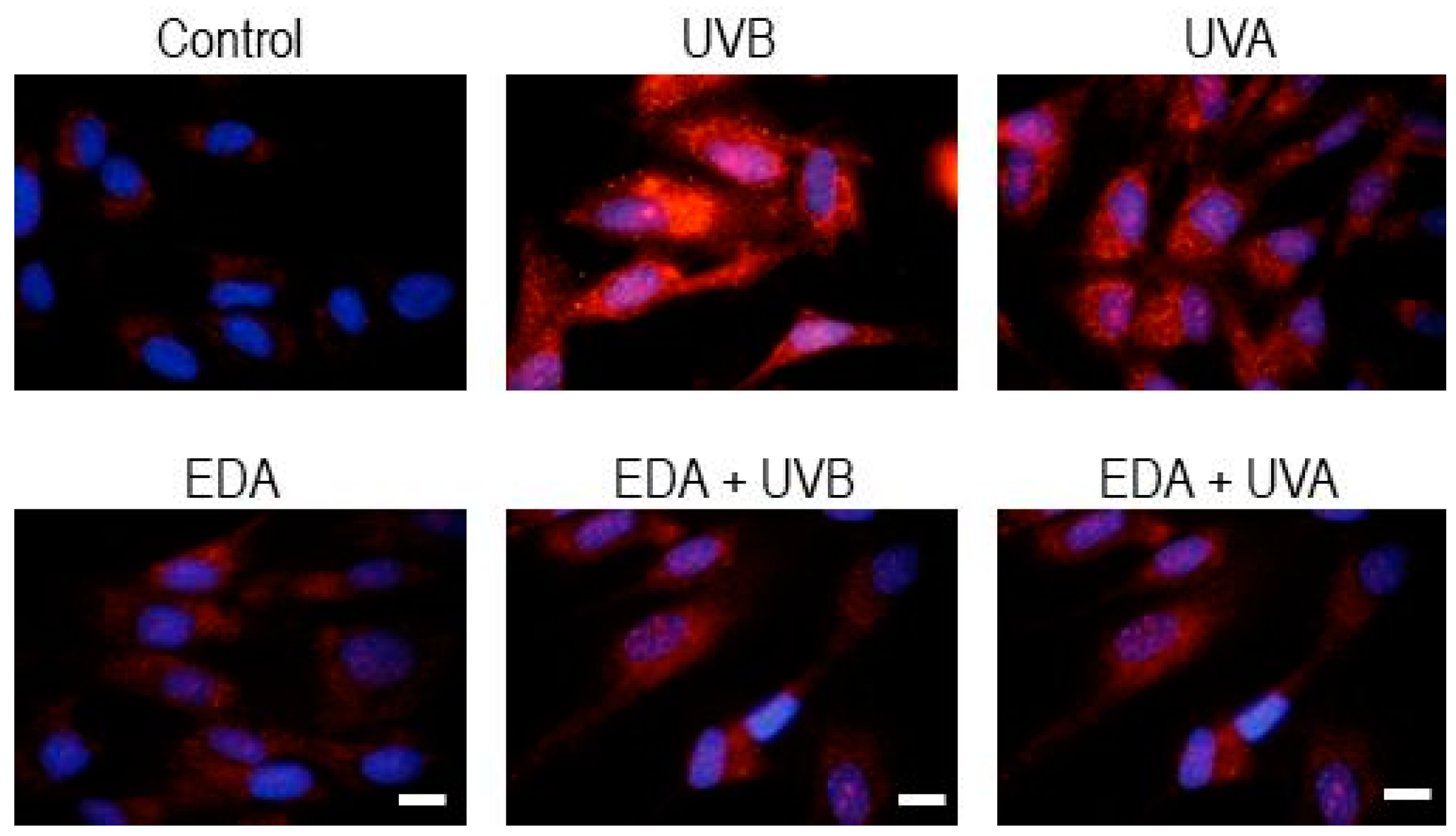
Human dermal fibroblast and keratinocytes were seeded on glass coverslips (Menzel-Gläser, Braunschweig, Germany) and when they reached an appropriate confluence (approximately 80%), they were incubated with EDA (0.5 mg/mL) for 24 h. Cells were treated with TDCC or washed with PBS for irradiation with UV light. After treatment, cells were fixed with 3.7% formaldehyde (Panreac, Barcelona, Spain), and then permeabilized with 0. 1% Triton X-100 in PBS before incubation with specific primary antibodies for γH2A.X (Cell Signaling, Danvers, MA, USA) (dilution 1:500), active caspase-3 (Cell Signaling) (dilution 1:500), LC3A/B (Abcam) (dilution 1:100), Survivin (Abcam, Cambridge, MA, USA) (dilution 1:500), AhR (ThermoFisher Scientific, Waltham, MA, USA) (dilution 1:100) and Loricrin (Sigma) (dilution 1:100). Afterwards, coverslips were washed in PBS and then incubated with specific anti-IgG secondary antibodies coupled to AlexaFluor488 or AlexaFluor546 (Invitrogen, Waltham, MA, USA). Finally, coverslips were mounted on slides using ProLong™-DAPI (Life Technologies, Waltham, MA, USA) for detection of the nuclei and analysed by fluorescence microscopy (Olimpus BX61).
4.7. Western Blot Assays
For the detection of nuclear AhR and laminin B (Dallas, TX, USA), the extract of the cytoplasmic and nuclear proteins was performed separately with the Nuclear Extraction Kit (Merk) according to manufacturer’s instructions. For the detection of total AhR, Tubulin, PARP and β-actin, extracts were obtained by using the Bio-world Protein Extraction Buffer with phosphatase inhibitors (Roche, Basel, Switzerland) and proteases (Roche), following the manufacturer’s instructions.
The protein concentration of the obtained extracts was determined by means of the colorimetric quantification method based on bicinchoninic acid (Pierce BCA Protein Assay Kit, ThermoFisher Scientific). Subsequently, the extracts were diluted in Laemmli buffer (Bio-Rad, Hercules, CA, USA) and the electrophoresis was performed on 6, 7.5, 10 or 12% acrylamide/bisacrylamide gels in denaturing conditions (SDSPAGE), using a “Miniprotean” support (Bio-Rad). 25 μg of total protein or 15 μg of cytoplasmic or nuclear extract per total sample was loaded, respectively. Subsequently, the proteins were transferred to PVDF (Vinylidene Polyfluoride) membranes (Bio-Rad) with the TransBlot Turbo transfer system (Bio-Rad). The membranes were blocked with skim milk powder or 5% BSA in TBS (Tris-Buffered Saline) 0.1% Tween® 20, according to the requirement of the antibody, for at least 1 h at room temperature under agitation. They were then incubated with the corresponding primary antibody anti-AhR (Thermo Fisher Scientific, Waltham, MA, USA) (dilution 1:250), anti-laminin B (Santa Cruz) (0.5 μg/mL), anti-tubulin (Sigma) (dilution 1:1000), anti-PARP (Cell Signaling) (dilution 1:500), and anti-β-actin (Santa Cruz Ltd.) (2 μg/mL) diluted in the same blocking solution overnight at 4° C under agitation, washed with 0.1% TBS-Tween® 20 and incubated with secondary mouse or rabbit antibody coupled to peroxidase (Thermo Fisher Scientific) for 2 h at room temperature. The development was carried out by chemiluminescence (ECL Plus kit, GE HealthcareChicago, IL, USA) using the ChemiDocTR XRS + high definition system (Bio-Rad). The bands corresponding to the different proteins were digitized using the Image Lab software version 3.0.1 (Bio-Rad).
4.8. Analysis of Cell Cycle
Near confluent HDF monolayers were trypsinized 24 and 48 h after treatment (EDA and/or UVB/A) and processed using a DNAprep kit (Qiagen, Hilden, Germany) according to manufacturer’s recommendations. Cellular cycle was evaluated by flow cytometry.
4.9. ELISA
MMP1 was analysed in supernatants from cultures of HDF treated with 0.1 or 0.3 mg/mL EDA for 24 h and then irradiated with UVB light at different dose: 20 and 40 mJ/cm2. Supernatants were collected 24 and 48 h post-treatment and MMP1 was analysed by ELISA assay (Becton & Dickinson, Franklin Lakes, NJ, USA).
4.10. Image Analysis
Microscopic images were obtained using an epifluorescence microscope coupled to a CCD camera DP70 (Olympus BX-61, Tokyo, Japan) with UV filters for the excitation light (360–370 nm excitation filter UG-1), blue (450–490 nm excitation filter BP 490), or green (570–590 nm excitation filter 590 DM). Picture processing was performed with Photoshop Extended CS5 12.0 (Adobe Systems Inc., Mountain View, CA, USA). Both percentages of positive cells and mean fluorescent intensity for specific antibodies were determined by using the Image J analysis software.
4.11. Statistical Analysis
Data are represented as the mean standard deviation (SD) of at least three independent experiments. Statistical significance was determined by statistical test of analysis of variance (ANOVA) or two-way ANOVA and Bonferroni post hoc tests, using GraphPad Prism 5.00 (GraphPad Software, Inc., San Diego, CA, USA). Differences were considered to be significant when p < 0.05.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








