Increased Expression of Vascular Endothelial Growth Factor-D Following Brain Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
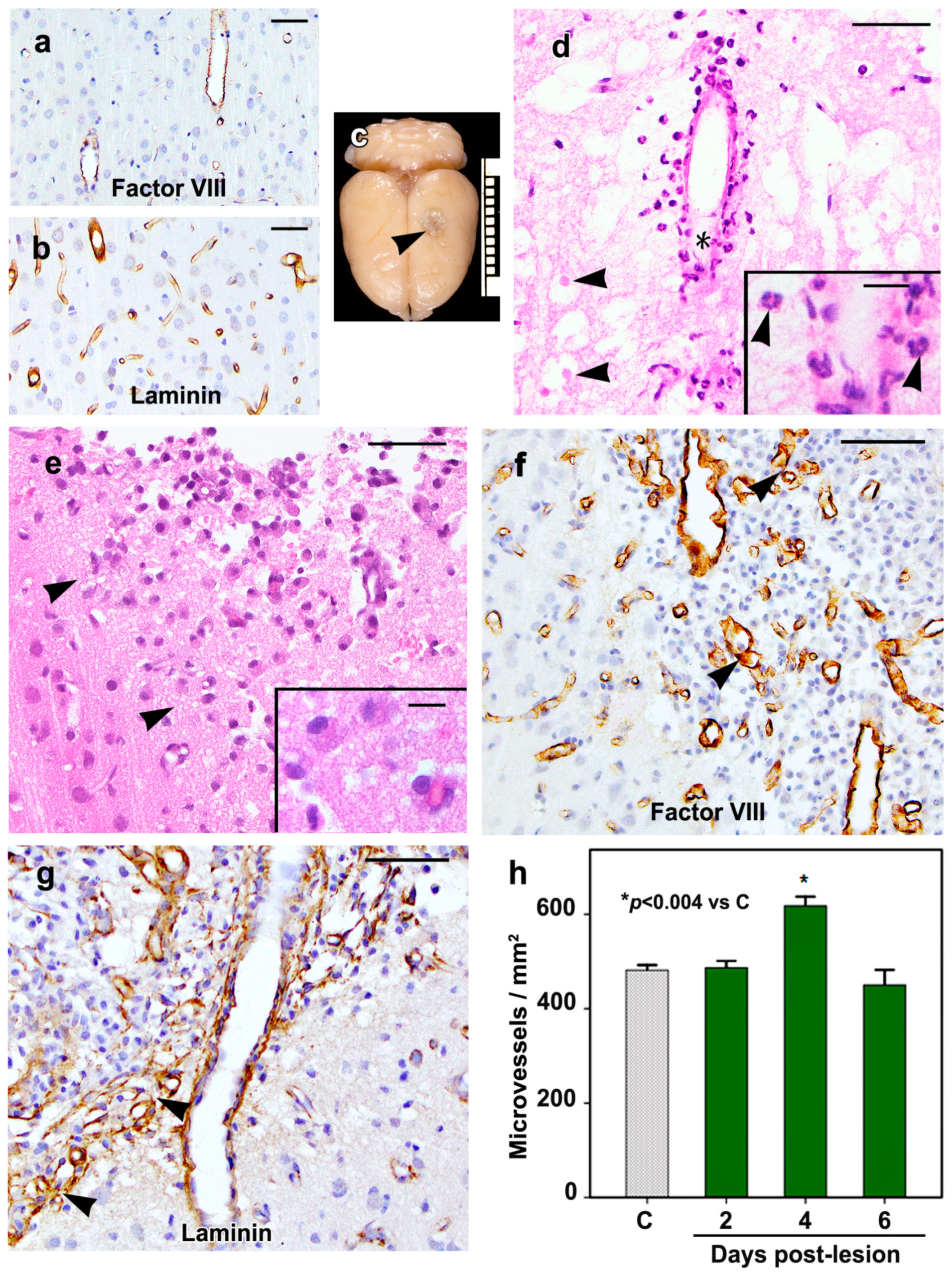
2.1. Morphology of the Cortical Cold Injury
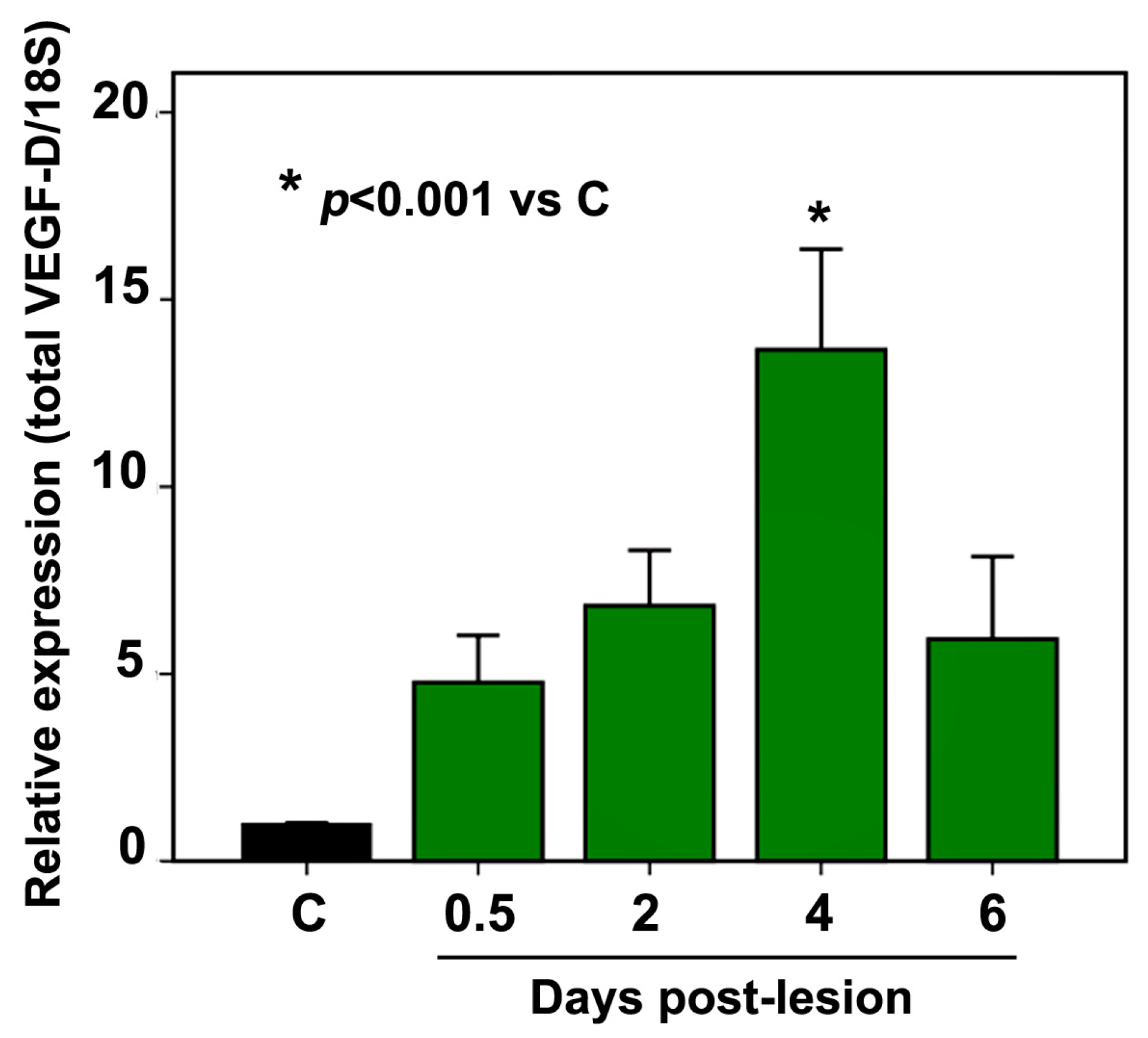
2.2. VEGF-D mRNA Expression
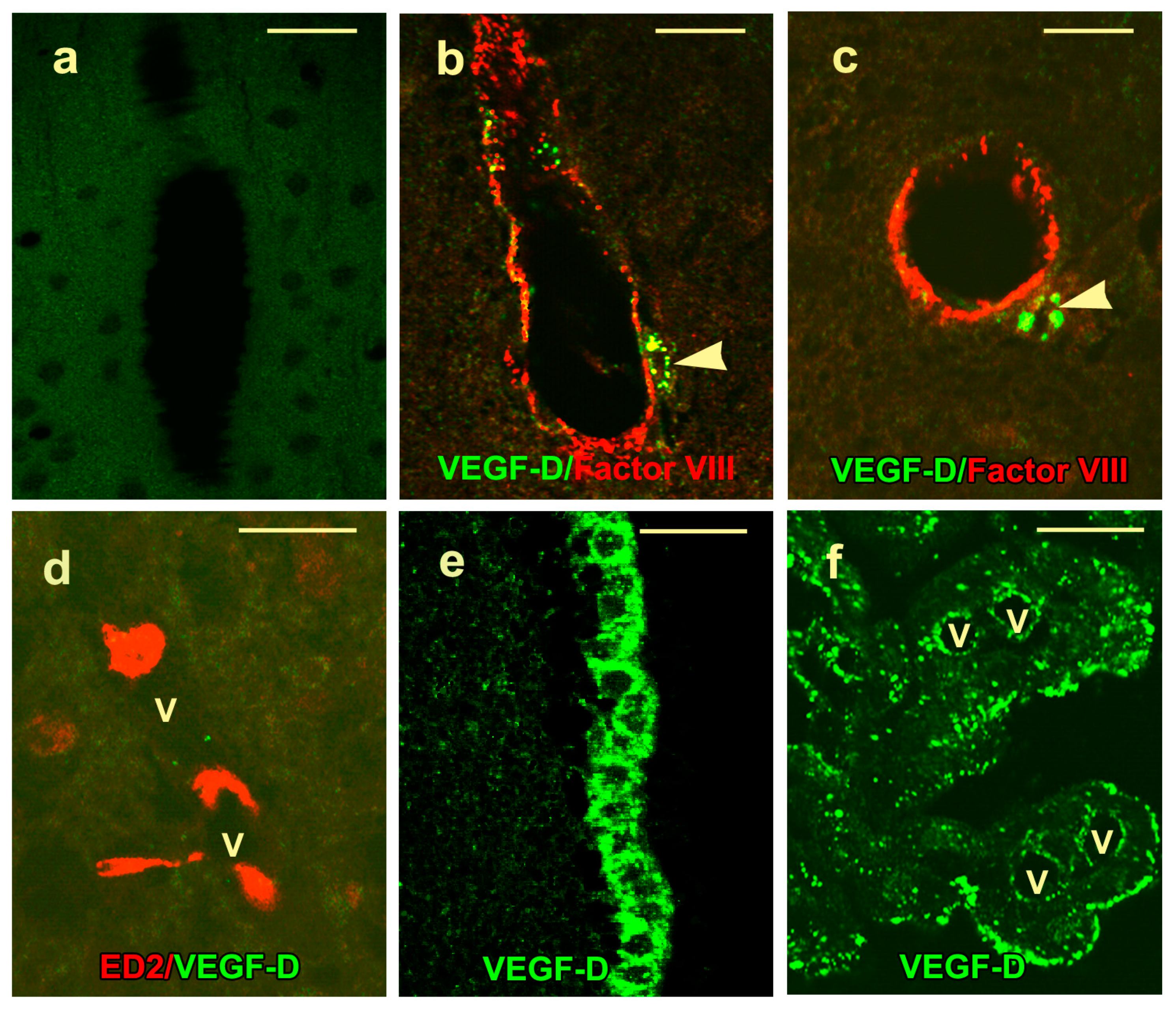
2.3. VEGF-D Protein Expression in Control Rats
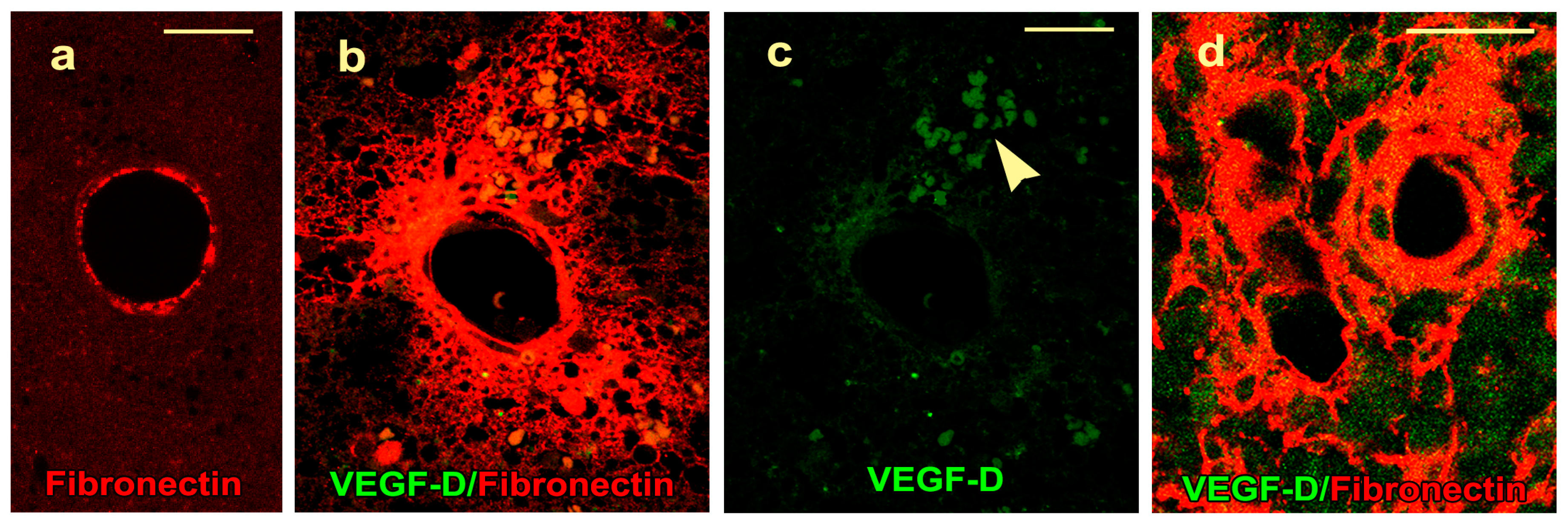
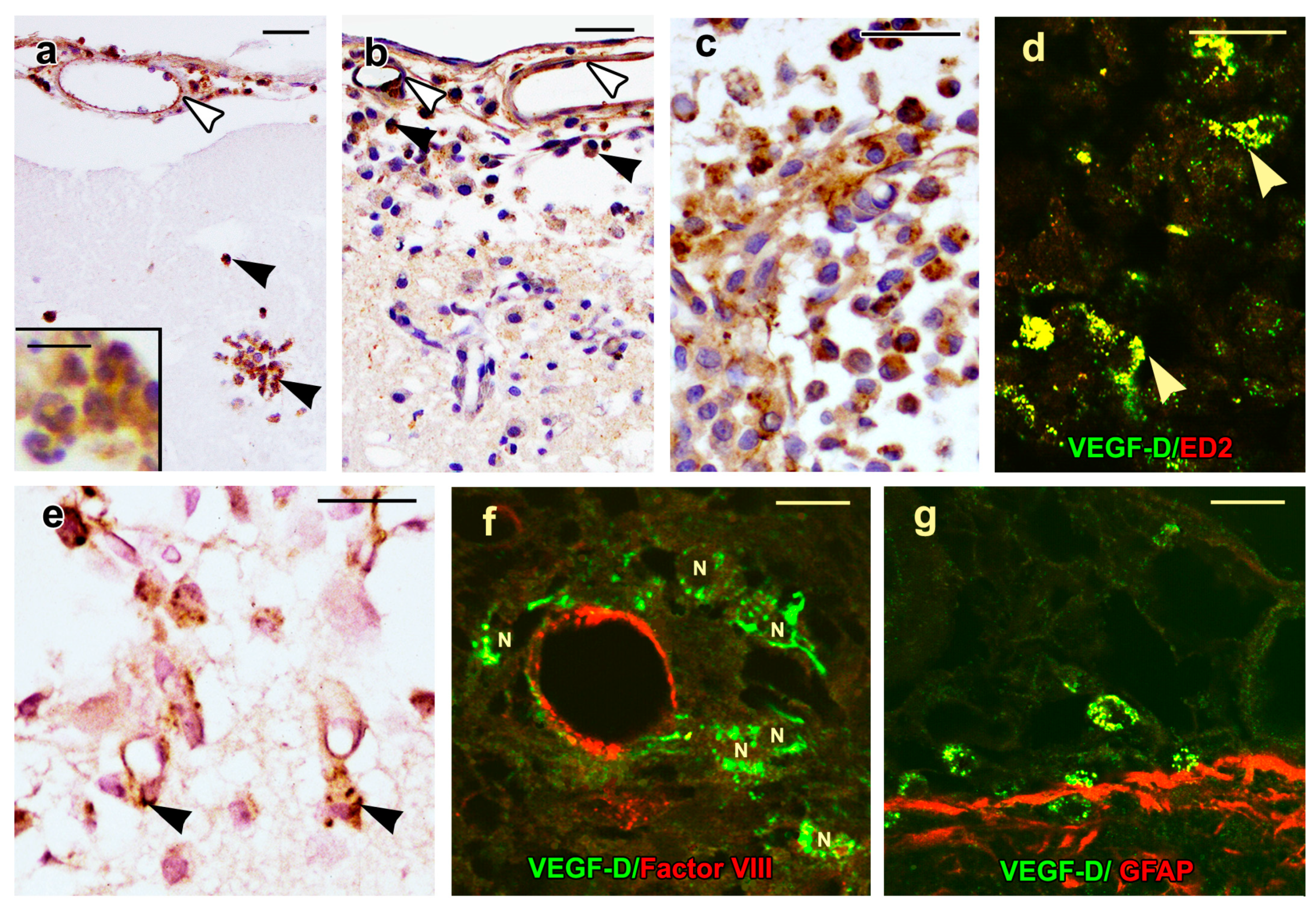
2.4. VEGF-D Protein Expression During BBB Breakdown
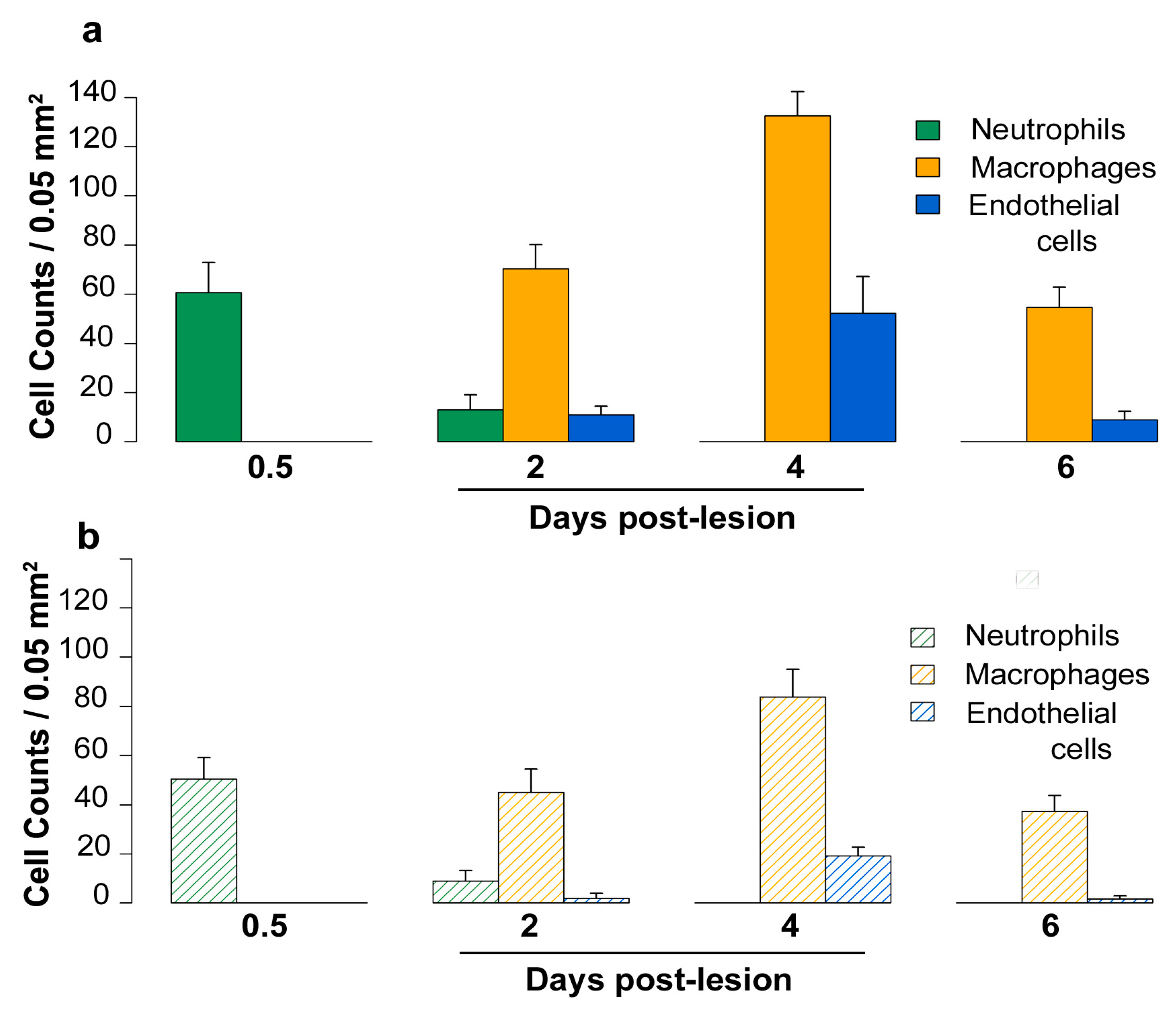
2.5. Density of Inflammatory and Free Endothelial Cells and Their Numbers Expressing VEGF-D Protein
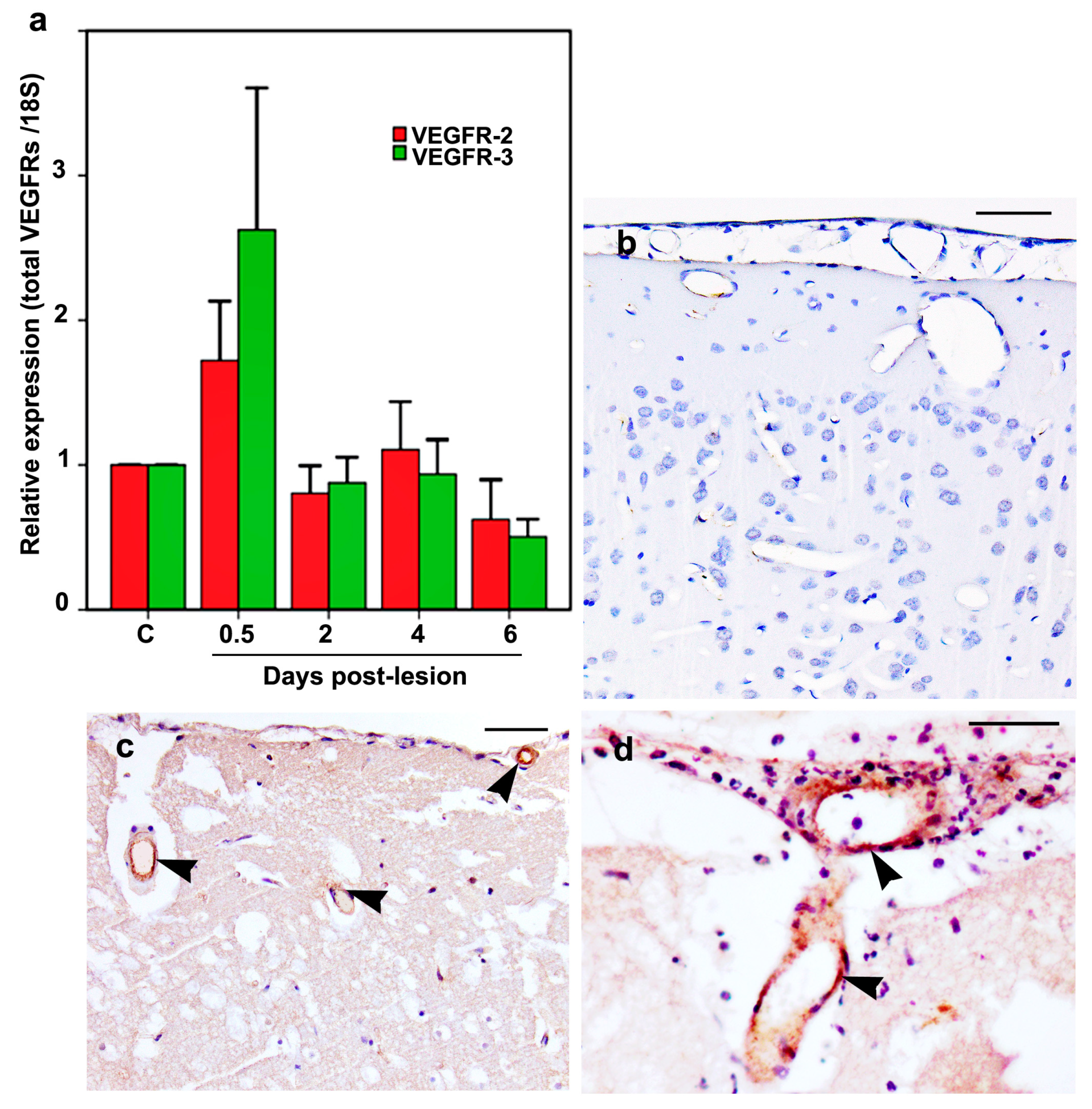
2.6. VEGFR-2 and VEGFR-3 mRNA and Protein Expression
3. Discussion
4. Materials and Methods
4.1. Cortical Cold-Injury Model
4.2. RNA Preparation and qRT-PCR
4.3. Histological Analyses and Immunohistochemistry
4.4. Immunofluorescence
4.5. Vessel and Cell Counts
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| ED2 | CD163 |
| HRP | Horseradish peroxidase |
| qRT-PCR | Quantitative reverse transcriptase polymerase chain reaction |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| 18S | 18S ribosomal RNA |
References
- Breier, G. Functions of the VEGF/VEGF receptor system in the vascular system. Semin. Thromb. Hemost. 2000, 26, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Jakeman, L.B.; Armanini, M.; Phillips, H.S.; Ferrara, N. Developmental expression of binding sites and messenger ribonucleic acid for vascular endothelial growth factor suggests a role for this protein in vasculogenesis and angiogenesis. Endocrinology 1993, 133, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Neufeld, G.; Gitay-Goren, H.; Keshet, E. Patterns of expression of vascular endothelial growth factor (VEGF) and VEGF receptors in mice suggest a role in hormonally regulated angiogenesis. J. Clin. Investig. 1993, 91, 2235–2243. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Hansen, T.M.; Moss, A.J.; Brindle, N.P. Vascular endothelial growth factor and angiopoietins in neurovascular regeneration and protection following stroke. Curr. Neurovasc. Res. 2008, 5, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Takahashi, J.L.; Kilty, D.W. Role of vascular endothelial growth factor in blood-brain barrier breakdown and angiogenesis in brain trauma. J. Neuropathol. Exp. Neurol. 1997, 56, 912–921. [Google Scholar] [CrossRef]
- Nag, S.; Eskandarian, M.R.; Davis, J.; Eubanks, J.H. Differential expression of vascular endothelial growth factor-A (VEGF-A) and VEGF-B after brain injury. J. Neuropathol. Exp. Neurol. 2002, 61, 778–788. [Google Scholar] [CrossRef]
- Salven, P.; Lymboussaki, A.; Heikkila, P.; JaaskelaSaari, H.; Enholm, B.; Aase, K.; von Euler, G.; Eriksson, U.; Alitalo, K.; Joensuu, H. Vascular endothelial growth factors VEGF-B and VEGF-C are expressed in human tumors. Am. J. Pathol. 1998, 153, 103–108. [Google Scholar] [CrossRef]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Makinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnebo, F.; Piehl, F.; Lagercrantz, J. Restricted expression pattern of vegf-d in the adult and fetal mouse: High expression in the embryonic lung. Biochem. Biophys. Res. Commun. 1999, 257, 891–894. [Google Scholar] [CrossRef]
- McColl, B.K.; Baldwin, M.E.; Roufail, S.; Freeman, C.; Moritz, R.L.; Simpson, R.J.; Alitalo, K.; Stacker, S.A.; Achen, M.G. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J. Exp. Med. 2003, 198, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Stenvers, K.; Caesar, C.; Vitali, A.; Domagala, T.; Nice, E.; Roufail, S.; Simpson, R.J.; Moritz, R.; Karpanen, T.; et al. Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J. Biol. Chem. 1999, 274, 32127–32136. [Google Scholar] [CrossRef] [PubMed]
- Avantaggiato, V.; Orlandini, M.; Acampora, D.; Oliviero, S.; Simeone, A. Embryonic expression pattern of the murine figf gene, a growth factor belonging to platelet-derived growth factor/vascular endothelial growth factor family. Mech. Dev. 1998, 73, 221–224. [Google Scholar] [CrossRef]
- Yamada, Y.; Nezu, J.; Shimane, M.; Hirata, Y. Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics 1997, 42, 483–488. [Google Scholar] [CrossRef]
- Baldwin, M.E.; Halford, M.M.; Roufail, S.; Williams, R.A.; Hibbs, M.L.; Grail, D.; Kubo, H.; Stacker, S.A.; Achen, M.G. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol. Cell. Biol. 2005, 25, 2441–2449. [Google Scholar] [CrossRef]
- Haiko, P.; Makinen, T.; Keskitalo, S.; Taipale, J.; Karkkainen, M.J.; Baldwin, M.E.; Stacker, S.A.; Achen, M.G.; Alitalo, K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol. Cell. Biol. 2008, 28, 4843–4850. [Google Scholar] [CrossRef]
- Achen, M.G.; Williams, R.A.; Minekus, M.P.; Thornton, G.E.; Stenvers, K.; Rogers, P.A.W.; Lederman, F.; Roufail, S.; Stacker, S.A. Localization of vascular endothelial growth factor-D in malignant melanoma suggests a role in tumour angiogenesis. J. Pathol. 2001, 193, 147–154. [Google Scholar] [CrossRef]
- Juttner, S.; Wissmann, C.; Jons, T.; Vieth, M.; Hertel, J.; Gretschel, S.; Schlag, P.M.; Kemmner, W.; Hocker, M. Vascular endothelial growth factor-D and its receptor VEGFR-3: Two novel independent prognostic markers in gastric adenocarcinoma. J. Clin. Oncol. 2006, 24, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Caesar, C.; Baldwin, M.E.; Thornton, G.E.; Williams, R.A.; Prevo, R.; Jackson, D.G.; Nishikawa, S.; Kubo, H.; Achen, M.G. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 2001, 7, 186–191. [Google Scholar] [CrossRef]
- Debinski, W.; Slagle-Webb, B.; Achen, M.G.; Stacker, S.A.; Tulchinsky, E.; Gillespie, G.Y.; Gibo, D.M. VEGF-D is an X-linked/AP-1 regulated putative onco-angiogen in human glioblastoma multiforme. Mol. Med. 2001, 7, 598–608. [Google Scholar] [CrossRef]
- Grau, S.J.; Trillsch, F.; Herms, J.; Thon, N.; Nelson, P.J.; Tonn, J.C.; Goldbrunner, R. Expression of VEGFR3 in glioma endothelium correlates with tumor grade. J. Neurooncol. 2007, 82, 141–150. [Google Scholar] [CrossRef]
- Klatzo, I.; Piraux, A.; Laskowski, E.J. The relationship between edema, blood-brain-barrier and tissue elements in a local brain injury. J. Neuropathol. Exp. Neurol. 1958, 17, 548–564. [Google Scholar] [CrossRef]
- Nag, S.; Manias, J.L.; Stewart, D.J. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. (Berl.) 2009, 118, 197–217. [Google Scholar] [CrossRef]
- Cancilla, P.A.; Frommes, S.P.; Kahn, L.E.; DeBault, L.E. Regeneration of cerebral microvessels: A morphologic and histochemical study after local freeze-injury. Lab. Investig. 1979, 40, 74–82. [Google Scholar]
- Nag, S. Cold-injury of the cerebral cortex: Immunolocalization of cellular proteins and blood-brain barrier permeability studies. J. Neuropathol. Exp. Neurol. 1996, 55, 880–888. [Google Scholar] [CrossRef]
- Nag, S. The blood-brain barrier and cerebral angiogenesis: Lessons from the cold-injury model. Trends Mol. Med. 2002, 8, 38–44. [Google Scholar] [CrossRef]
- Nag, S.; Venugopalan, R.; Stewart, D.J. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. (Berl.) 2007, 114, 459–469. [Google Scholar] [CrossRef]
- Nag, S.; Manias, J.L.; Kapadia, A.; Stewart, D.J. Molecular Changes Associated with the Protective Effects of Angiopoietin-1 During Blood-Brain Barrier Breakdown Post-Injury. Mol. Neurobiol. 2017, 54, 4232–4242. [Google Scholar] [CrossRef]
- Nag, S.; Picard, P.; Stewart, D.J. Expression of nitric oxide synthases and nitrotyrosine during blood-brain barrier breakdown and repair after cold injury. Lab. Investig. 2001, 81, 41–49. [Google Scholar] [CrossRef]
- Nag, S. Blood-brain barrier permeability using tracers and immunohistochemistry. Methods Mol. Med. 2003, 89, 133–144. [Google Scholar]
- Dallasta, L.M.; Pisarov, L.A.; Esplen, J.E.; Werley, J.V.; Moses, A.V.; Nelson, J.A.; Achim, C.L. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 1999, 155, 915–1927. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Nagy, J.A.; Feng, D.; Brown, L.F.; Dvorak, A.M. Vascular permeability factor vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Am. J. Pathol. 1999, 146, 97–132. [Google Scholar]
- Orlandini, M.; Marconcini, L.; Ferruzzi, R.; Oliviero, S. Identification of a c-fos-induced gene that is related to the platelet-derived growth factor/vascular endothelial growth factor family. Proc. Natl. Acad. Sci. USA 1996, 93, 11675–11680. [Google Scholar] [CrossRef]
- Kopfstein, L.; Veikkola, T.; Djonov, V.G.; Baeriswyl, V.; Schomber, T.; Strittmatter, K.; Stacker, S.A.; Achen, M.G.; Alitalo, K.; Christofori, G. Distinct roles of vascular endothelial growth factor-D in lymphangiogenesis and metastasis. Am. J. Pathol. 2007, 170, 1348–1361. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Roy, H.; Gruchala, M.; Viita, H.; Kholova, I.; Kokina, I.; Achen, M.G.; Stacker, S.A.; Hedman, M.; Alitalo, K.; et al. Angiogenic responses of vascular endothelial growth factors in periadventitial tissue. Hum. Gene Ther. 2003, 14, 1451–1462. [Google Scholar] [CrossRef]
- Rissanen, T.T.; Markkanen, J.E.; Gruchala, M.; Heikura, T.; Puranen, A.; Kettunen, M.I.; Kholova, I.; Kauppinen, R.A.; Achen, M.G.; Stacker, S.A.; et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ. Res. 2003, 92, 1098–1106. [Google Scholar] [CrossRef]
- Millauer, B.; Wizigmann-Voos, S.; Schnurch, H.; Martinez, R.; Moller, N.P.; Risau, W.; Ullrich, A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72, 835–846. [Google Scholar] [CrossRef]
- Kaipainen, A.; Korhonen, J.; Mustonen, T.; Van Hinsbergh, V.W.; Fang, G.H.; Dumont, D.; Breitman, M.; Alitalo, K. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc. Natl. Acad. Sci. USA 1995, 92, 3566–3570. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Nurmi, H.; Jakobsson, L.; Heinolainen, K.; Tvorogov, D.; Zheng, W.; Franco, C.A.; Murtomaki, A.; Aranda, E.; et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat. Cell Biol. 2011, 13, 1202–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Blood-brain barrier genomics. Stroke 2007, 38 (Suppl. 2), 686–690. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Breier, G.; Millauer, B.; Ullrich, A.; Risau, W. Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res. 1993, 53, 5822–5827. [Google Scholar]
- Skold, M.K.; Gertten, C.V.; Sandberg-Nordqvist, A.C.; Mathiesen, T.; Holmin, S. VEGF and VEGF receptor expression after experimental brain contusion in rat. J. Neurotrauma 2005, 22, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Krum, J.M.; Mani, N.; Rosenstein, J.M. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience 2002, 110, 589–604. [Google Scholar] [CrossRef]
- Lafuente, J.V.; Argandona, E.G.; Mitre, B. VEGFR-2 expression in brain injury: Its distribution related to brain-blood barrier markers. J. Neural Transm. (Vienna) 2006, 113, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Chodobski, A.; Chung, I.; Kozniewska, E.; Ivanenko, T.; Chang, W.; Harrington, J.F.; Duncan, J.A.; Szmydynger-Chodobska, J. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience 2003, 122, 853–867. [Google Scholar] [CrossRef]
- Dijkstra, C.D.; Dopp, E.A.; Joling, P.; Kraal, G. The heterogeneity of mononuclear phagocytes in lymphoid organs: Distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology 1985, 54, 589–599. [Google Scholar]
- Graeber, M.B.; Streit, W.J.; Kreutzberg, G.W. Identity of ED2-positive perivascular cells in rat brain. J. Neurosci. Res. 1989, 22, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Nag, S. Morphology and molecular properties of cellular components of normal cerebral vessels. Methods Mol. Med. 2003, 89, 3–36. [Google Scholar]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Moffat, B.A.; Chen, M.; Kariaapper, M.S.; Hamstra, D.A.; Hall, D.E.; Stojanovska, J.; Johnson, T.D.; Blaivas, M.; Kumar, M.; Chenevert, T.L.; et al. Inhibition of vascular endothelial growth factor (VEGF)-A causes a paradoxical increase in tumor blood flow and up-regulation of VEGF-D. Clin. Cancer Res. 2006, 12, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Achen, M.G. Emerging Roles for VEGF-D in Human Disease. Biomolecules 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Mauceri, D.; Freitag, H.E.; Oliveira, A.M.; Bengtson, C.P.; Bading, H. Nuclear calcium-VEGFD signaling controls maintenance of dendrite arborization necessary for memory formation. Neuron 2011, 71, 117–130. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sena, K.; Sumner, D.R.; Virdi, A.S. Modulation of VEGF expression in rat bone marrow stromal cells by GDF-5. Connect. Tissue Res. 2007, 48, 324–331. [Google Scholar] [CrossRef] [PubMed]
- McCarter, S.D.; Mei, S.H.; Lai, P.F.; Zhang, Q.W.; Parker, C.H.; Suen, R.S.; Hood, R.D.; Zhao, Y.D.; Deng, Y.; Han, R.N.; et al. Cell-based angiopoietin-1 gene therapy for acute lung injury. Am. J. Respir. Crit. Care Med. 2007, 175, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Manias, J.L.; Kapadia, A.; Nag, S. Detection of multiple proteins in intracerebral vessels by confocal microscopy. Methods Mol. Biol. 2011, 686, 177–192. [Google Scholar]
- Nag, S.; Manias, J.L.; Stewart, D.J. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol. Appl. Neurobiol. 2009, 35, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sharma, M.C.; Sarkar, C. Morphology of angiogenesis in human cancer: A conceptual overview, histoprognostic perspective and significance of neoangiogenesis. Histopathology 2005, 46, 481–489. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nag, S.; Manias, J.; Eubanks, J.H.; Stewart, D.J. Increased Expression of Vascular Endothelial Growth Factor-D Following Brain Injury. Int. J. Mol. Sci. 2019, 20, 1594. https://doi.org/10.3390/ijms20071594
Nag S, Manias J, Eubanks JH, Stewart DJ. Increased Expression of Vascular Endothelial Growth Factor-D Following Brain Injury. International Journal of Molecular Sciences. 2019; 20(7):1594. https://doi.org/10.3390/ijms20071594
Chicago/Turabian StyleNag, Sukriti, Janet Manias, James H. Eubanks, and Duncan J. Stewart. 2019. "Increased Expression of Vascular Endothelial Growth Factor-D Following Brain Injury" International Journal of Molecular Sciences 20, no. 7: 1594. https://doi.org/10.3390/ijms20071594
APA StyleNag, S., Manias, J., Eubanks, J. H., & Stewart, D. J. (2019). Increased Expression of Vascular Endothelial Growth Factor-D Following Brain Injury. International Journal of Molecular Sciences, 20(7), 1594. https://doi.org/10.3390/ijms20071594