Molecular Mechanisms of Kidney Injury and Repair in Arterial Hypertension


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical and Histopathological Characteristics of Kidney Injury in Arterial Hypertension
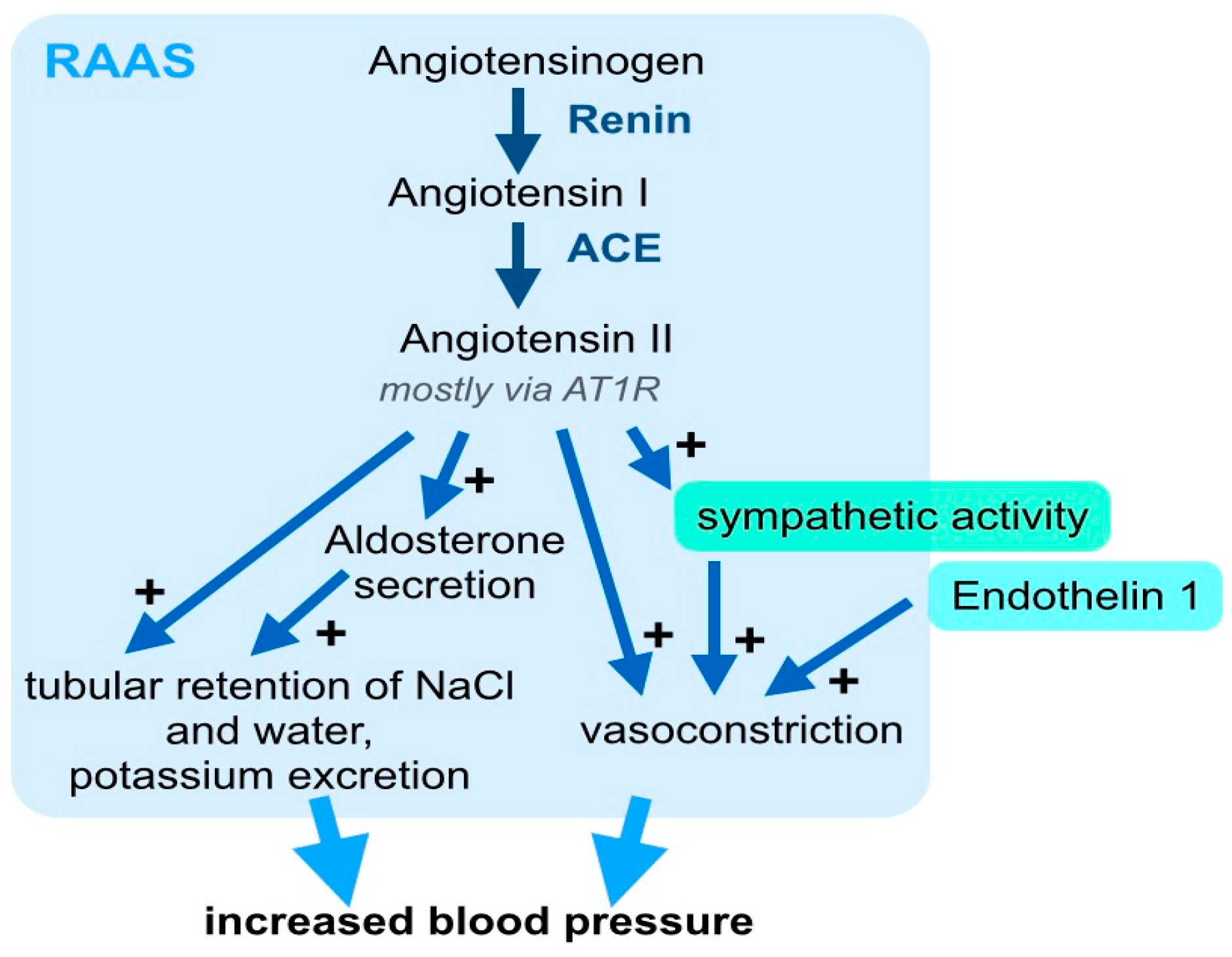
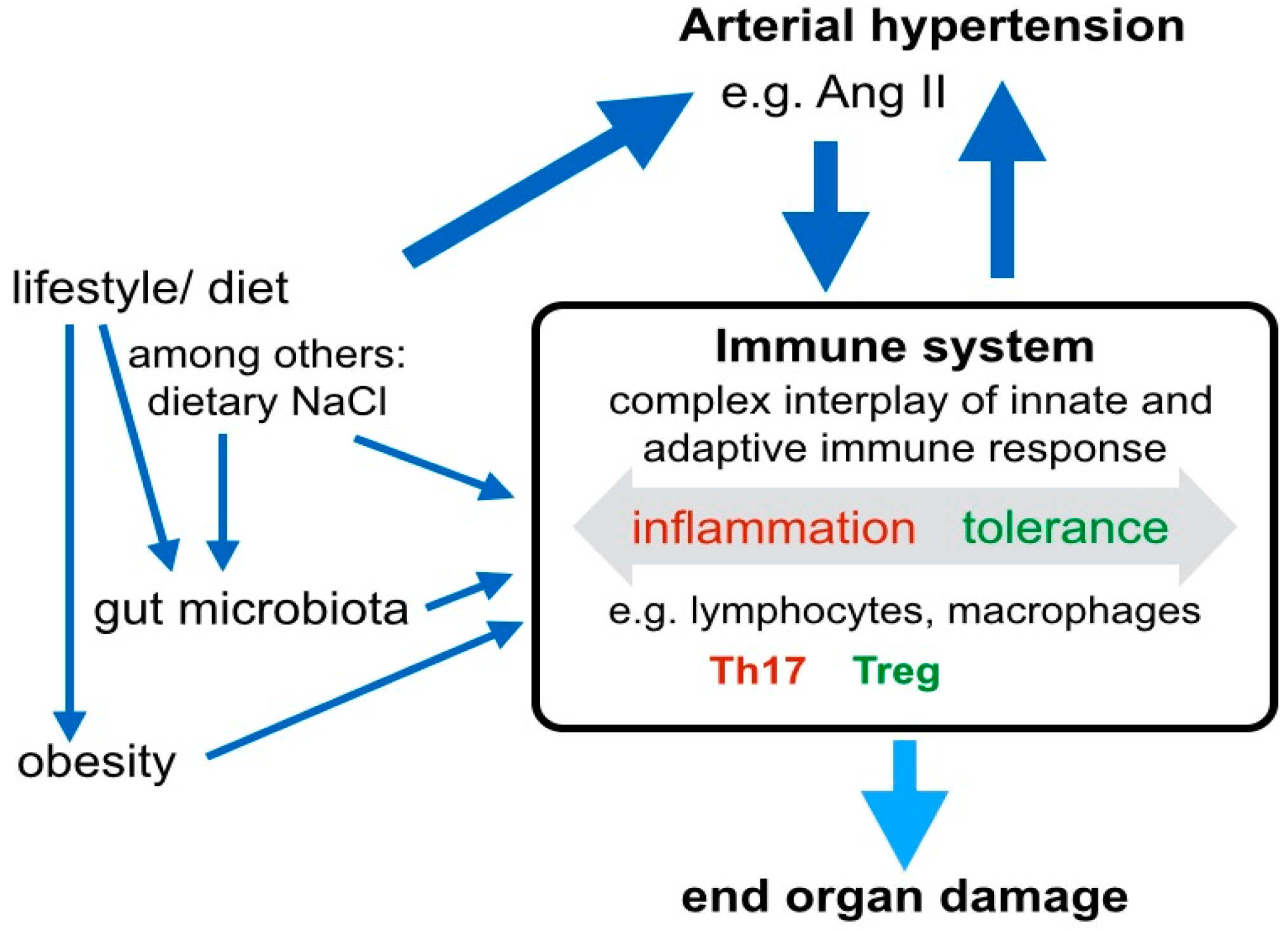
3. Systemic Signaling Pathways and the Immune System in Arterial Hypertension
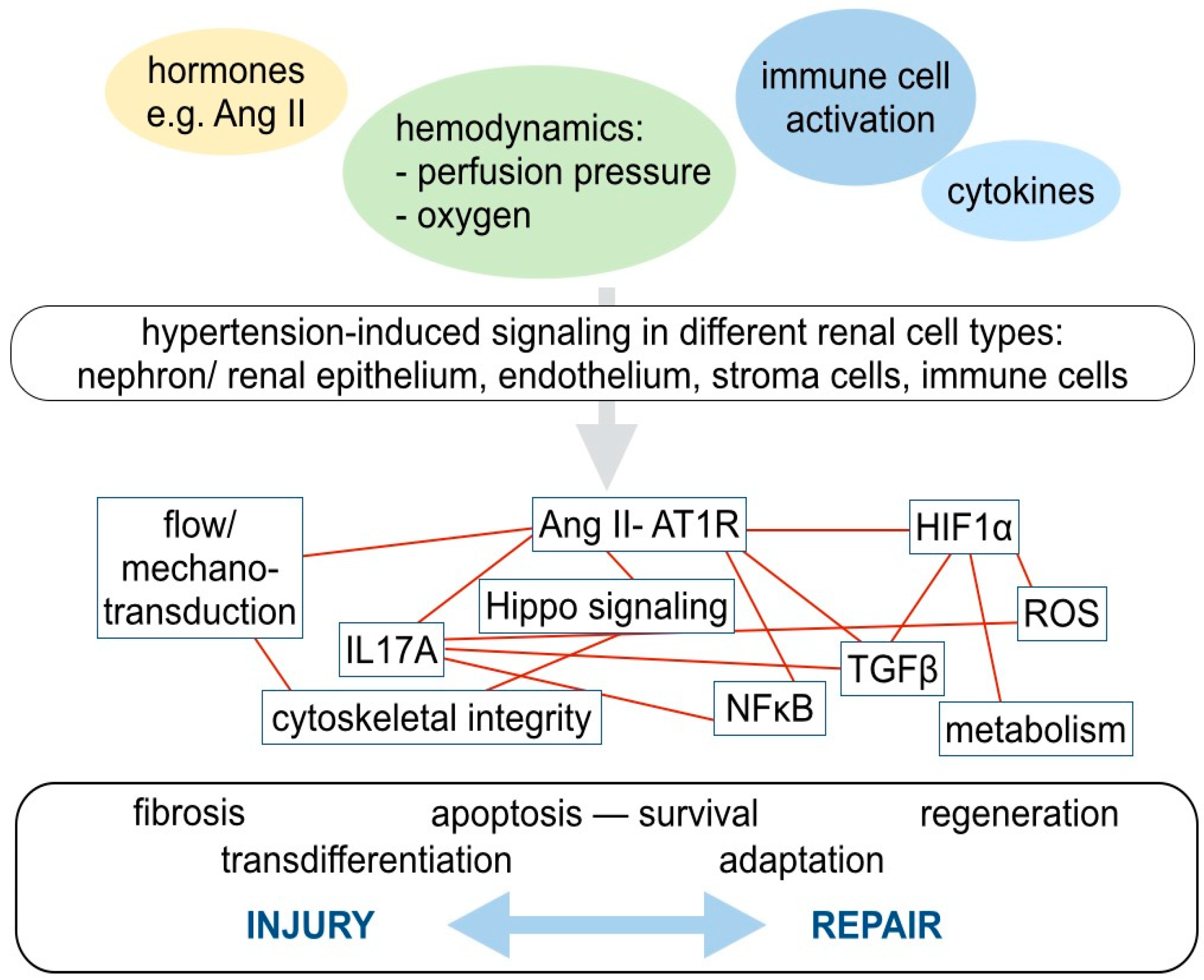
4. Molecular Mechanisms in Hypertensive Kidney Injury
5. Approaches to Foster Regeneration and Repair in Hypertensive Nephropathy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scheppach, J.B.; Raff, U.; Toncar, S.; Ritter, C.; Klink, T.; Stork, S.; Wanner, C.; Schlieper, G.; Saritas, T.; Reinartz, S.D.; et al. Blood Pressure Pattern and Target Organ Damage in Patients with Chronic Kidney Disease. Hypertension 2018, 72, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Le, W.; Liang, D.; Chen, H.; Xu, F.; Chen, H.; Liu, Z.; Zeng, C. Clinico-pathological characteristics and outcomes of patients with biopsy-proven hypertensive nephrosclerosis: A retrospective cohort study. BMC Nephrol. 2016, 17, 42. [Google Scholar] [CrossRef]
- Hill, G.S. Hypertensive nephrosclerosis. Curr. Opin. Nephrol. Hypertens. 2008, 17, 266–270. [Google Scholar] [CrossRef]
- Thomas, J.L.; Pham, H.; Li, Y.; Hall, E.; Perkins, G.A.; Ali, S.S.; Patel, H.H.; Singh, P. Hypoxia-inducible factor-1alpha activation improves renal oxygenation and mitochondrial function in early chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2017, 313, F282–F290. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The Force that Drives Chronic Kidney Disease. Clin. Med. Res. 2016, 14, 15–39. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M.; Rosenberger, C.; Heyman, S.N.; Eckardt, K.U. Regulation of hypoxia-inducible factor in kidney disease. Clin. Exp. Pharmacol. Physiol. 2013, 40, 148–157. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Reboussin, D.M.; Allen, N.B.; Griswold, M.E.; Guallar, E.; Hong, Y.; Lackland, D.T.; Miller, E.P.R., 3rd.; Polonsky, T.; Thompson-Paul, A.M.; Vupputuri, S. Systematic Review for the 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2018, 138, e595–e616. [Google Scholar]
- Jordan, J. Device-Based Approaches for the Treatment of Arterial Hypertension. Curr. Hypertens. Rep. 2017, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Nadim, M.K.; Haller, H.; Lovett, E.G.; Schafer, J.E.; Bisognano, J.D. Baroreflex activation therapy provides durable benefit in patients with resistant hypertension: Results of long-term follow-up in the Rheos Pivotal Trial. J. Am. Soc. Hypertens. 2012, 6, 152–158. [Google Scholar] [CrossRef]
- Singh, R.R.; Denton, K.M. Renal Denervation. Hypertension 2018, 72, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Persu, A.; Kjeldsen, S.; Staessen, J.A.; Azizi, M. Renal Denervation for Treatment of Hypertension: A Second Start and New Challenges. Curr. Hypertens. Rep. 2016, 18, 6. [Google Scholar] [CrossRef]
- Symplicity HTN-2 Investigators; Esler, M.D.; Krum, H.; Sobotka, P.A.; Schlaich, M.P.; Schmieder, R.E.; Bohm, M. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial): A randomised controlled trial. Lancet 2010, 376, 1903–1909. [Google Scholar]
- Mahfoud, F.; Bohm, M.; Schmieder, R.; Narkiewicz, K.; Ewen, S.; Ruilope, L.; Schlaich, M.; Williams, B.; Fahy, M.; Mancia, G. Effects of renal denervation on kidney function and long-term outcomes: 3-year follow-up from the Global SYMPLICITY Registry. Eur. Heart J. 2019. [Google Scholar] [CrossRef]
- Townsend, R.R.; Mahfoud, F.; Kandzari, D.E.; Kario, K.; Pocock, S.; Weber, M.A.; Ewen, S.; Tsioufis, K.; Tousoulis, D.; Sharp, A.S.P.; et al. Catheter-based renal denervation in patients with uncontrolled hypertension in the absence of antihypertensive medications (SPYRAL HTN-OFF MED): A randomised, sham-controlled, proof-of-concept trial. Lancet 2017, 390, 2160–2170. [Google Scholar] [CrossRef]
- Azizi, M.; Schmieder, R.E.; Mahfoud, F.; Weber, M.A.; Daemen, J.; Davies, J.; Basile, J.; Kirtane, A.J.; Wang, Y.; Lobo, M.D.; et al. Endovascular ultrasound renal denervation to treat hypertension (RADIANCE-HTN SOLO): A multicentre, international, single-blind, randomised, sham-controlled trial. Lancet 2018, 391, 2335–2345. [Google Scholar] [CrossRef]
- Kandzari, D.E.; Bohm, M.; Mahfoud, F.; Townsend, R.R.; Weber, M.A.; Pocock, S.; Tsioufis, K.; Tousoulis, D.; Choi, J.W.; East, C.; et al. Effect of renal denervation on blood pressure in the presence of antihypertensive drugs: 6-month efficacy and safety results from the SPYRAL HTN-ON MED proof-of-concept randomised trial. Lancet 2018, 391, 2346–2355. [Google Scholar] [CrossRef]
- Mian, M.O.; Barhoumi, T.; Briet, M.; Paradis, P.; Schiffrin, E.L. Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. J. Hypertens. 2016, 34, 97–108. [Google Scholar] [CrossRef]
- Barhoumi, T.; Kasal, D.A.; Li, M.W.; Shbat, L.; Laurant, P.; Neves, M.F.; Paradis, P.; Schiffrin, E.L. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 2011, 57, 469–476. [Google Scholar] [CrossRef]
- Mehrotra, P.; Patel, J.B.; Ivancic, C.M.; Collett, J.A.; Basile, D.P. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. 2015, 88, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Lee, S.H.; Lee, A.; Kim, D.J.; Kim, Y.G.; Kim, S.Y.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Lim, S.J.; et al. Targeting T helper 17 by mycophenolate mofetil attenuates diabetic nephropathy progression. Transl. Res. 2015, 166, 375–383. [Google Scholar] [CrossRef]
- Mohamed, R.; Jayakumar, C.; Chen, F.; Fulton, D.; Stepp, D.; Gansevoort, R.T.; Ramesh, G. Low-Dose IL-17 Therapy Prevents and Reverses Diabetic Nephropathy, Metabolic Syndrome, and Associated Organ Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 745–765. [Google Scholar] [CrossRef]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mahler, A.; Balogh, A.; Marko, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Marko, L.; Hoges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. The Short-Chain Fatty Acid Propionate Protects from Hypertensive Cardiovascular Damage. Circulation 2018. [Google Scholar] [CrossRef] [PubMed]
- Higaki, A.; Caillon, A.; Paradis, P.; Schiffrin, E.L. Innate and Innate-Like Immune System in Hypertension and Vascular Injury. Curr. Hypertens. Rep. 2019, 21, 4. [Google Scholar] [CrossRef]
- Warren, H.R.; Evangelou, E.; Cabrera, C.P.; Gao, H.; Ren, M.; Mifsud, B.; Ntalla, I.; Surendran, P.; Liu, C.; Cook, J.P.; et al. Corrigendum: Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat. Genet. 2017, 49, 1558. [Google Scholar] [CrossRef] [PubMed]
- Delles, C.; Carrick, E.; Graham, D.; Nicklin, S.A. Utilizing proteomics to understand and define hypertension: Where are we and where do we go? Expert Rev. Proteomics 2018, 15, 581–592. [Google Scholar] [CrossRef]
- Giani, J.F.; Janjulia, T.; Taylor, B.; Bernstein, E.A.; Shah, K.; Shen, X.Z.; McDonough, A.A.; Bernstein, K.E.; Gonzalez-Villalobos, R.A. Renal generation of angiotensin II and the pathogenesis of hypertension. Curr. Hypertens. Rep. 2014, 16, 477. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef]
- Fukuda, A.; Wickman, L.T.; Venkatareddy, M.P.; Sato, Y.; Chowdhury, M.A.; Wang, S.Q.; Shedden, K.A.; Dysko, R.C.; Wiggins, J.E.; Wiggins, R.C. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int. 2012, 81, 40–55. [Google Scholar] [CrossRef]
- Gloy, J.; Henger, A.; Fischer, K.G.; Nitschke, R.; Mundel, P.; Bleich, M.; Schollmeyer, P.; Greger, R.; Pavenstadt, H. Angiotensin II depolarizes podocytes in the intact glomerulus of the Rat. J. Clin. Invest. 1997, 99, 2772–2781. [Google Scholar] [CrossRef]
- Henger, A.; Huber, T.; Fischer, K.G.; Nitschke, R.; Mundel, P.; Schollmeyer, P.; Greger, R.; Pavenstadt, H. Angiotensin II increases the cytosolic calcium activity in rat podocytes in culture. Kidney Int. 1997, 52, 687–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijenhuis, T.; Sloan, A.J.; Hoenderop, J.G.; Flesche, J.; van Goor, H.; Kistler, A.D.; Bakker, M.; Bindels, R.J.; de Boer, R.A.; Moller, C.C.; et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am. J. Pathol. 2011, 179, 1719–1732. [Google Scholar] [CrossRef]
- Schenk, L.K.; Moller-Kerutt, A.; Klosowski, R.; Wolters, D.; Schaffner-Reckinger, E.; Weide, T.; Pavenstadt, H.; Vollenbroker, B. Angiotensin II regulates phosphorylation of actin-associated proteins in human podocytes. FASEB J. 2017, 31, 5019–5035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Liang, W.; Chen, C.; Yang, H.; Singhal, P.C.; Ding, G. Angiotensin II induces nephrin dephosphorylation and podocyte injury: Role of caveolin-1. Cell Signal. 2012, 24, 443–450. [Google Scholar] [CrossRef]
- Yang, Q.; Ma, Y.; Liu, Y.; Liang, W.; Chen, X.; Ren, Z.; Wang, H.; Singhal, P.C.; Ding, G. Angiotensin II down-regulates nephrin-Akt signaling and induces podocyte injury: Roleof c-Abl. Mol. Biol. Cell 2016, 27, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Wennmann, D.O.; Vollenbroker, B.; Eckart, A.K.; Bonse, J.; Erdmann, F.; Wolters, D.A.; Schenk, L.K.; Schulze, U.; Kremerskothen, J.; Weide, T.; et al. The Hippo pathway is controlled by Angiotensin II signaling and its reactivation induces apoptosis in podocytes. Cell Death Dis. 2014, 5, e1519. [Google Scholar] [CrossRef]
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Q.; Yang, J.; Ma, Y.; Ding, G. Angiotensin II induces cholesterol accumulation and injury in podocytes. Sci. Rep. 2017, 7, 10672. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Youssef, S.; Hur, E.M.; Ho, P.P.; Han, M.H.; Lanz, T.V.; Phillips, L.K.; Goldstein, M.J.; Bhat, R.; Raine, C.S.; et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2009, 106, 14948–14953. [Google Scholar] [CrossRef] [Green Version]
- Caillon, A.; Mian, M.O.R.; Fraulob-Aquino, J.C.; Huo, K.G.; Barhoumi, T.; Ouerd, S.; Sinnaeve, P.R.; Paradis, P.; Schiffrin, E.L. gammadelta T Cells Mediate Angiotensin II-Induced Hypertension and Vascular Injury. Circulation 2017, 135, 2155–2162. [Google Scholar] [CrossRef]
- Ruster, C.; Wolf, G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J. Am. Soc. Nephrol. 2011, 22, 1189–1199. [Google Scholar] [CrossRef]
- Suthanthiran, M.; Li, B.; Song, J.O.; Ding, R.; Sharma, V.K.; Schwartz, J.E.; August, P. Transforming growth factor-beta 1 hyperexpression in African-American hypertensives: A novel mediator of hypertension and/or target organ damage. Proc. Natl. Acad. Sci. USA 2000, 97, 3479–3484. [Google Scholar] [CrossRef]
- Suthanthiran, M.; Gerber, L.M.; Schwartz, J.E.; Sharma, V.K.; Medeiros, M.; Marion, R.; Pickering, T.G.; August, P. Circulating transforming growth factor-beta1 levels and the risk for kidney disease in African Americans. Kidney Int. 2009, 76, 72–80. [Google Scholar] [CrossRef]
- Feng, Y.; Liang, Y.; Zhu, X.; Wang, M.; Gui, Y.; Lu, Q.; Gu, M.; Xue, X.; Sun, X.; He, W.; et al. The signaling protein Wnt5a promotes TGFbeta1-mediated macrophage polarization and kidney fibrosis by inducing the transcriptional regulators Yap/Taz. J. Biol. Chem. 2018, 293, 19290–19302. [Google Scholar] [CrossRef]
- Vigolo, E.; Marko, L.; Hinze, C.; Muller, D.N.; Schmidt-Ullrich, R.; Schmidt-Ott, K.M. Canonical BMP signaling in tubular cells mediates recovery after acute kidney injury. Kidney Int. 2019, 95, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Brinks, H.L.; Eckhart, A.D. Regulation of GPCR signaling in hypertension. Biochim. Biophys. Acta 2010, 1802, 1268–1275. [Google Scholar] [CrossRef]
- Yang, J.; Villar, V.A.; Armando, I.; Jose, P.A.; Zeng, C. G Protein-Coupled Receptor Kinases: Crucial Regulators of Blood Pressure. J. Am. Heart Assoc. 2016, 5, e003519. [Google Scholar] [CrossRef] [PubMed]
- Kamal, F.A.; Travers, J.G.; Schafer, A.E.; Ma, Q.; Devarajan, P.; Blaxall, B.C. G Protein-Coupled Receptor-G-Protein betagamma-Subunit Signaling Mediates Renal Dysfunction and Fibrosis in Heart Failure. J. Am. Soc. Nephrol. 2017, 28, 197–208. [Google Scholar] [CrossRef]
- Deb, D.K.; Bao, R.; Li, Y.C. Critical role of the cAMP-PKA pathway in hyperglycemia-induced epigenetic activation of fibrogenic program in the kidney. FASEB J. 2017, 31, 2065–2075. [Google Scholar] [CrossRef]
- Luo, R.; Zhang, W.; Zhao, C.; Zhang, Y.; Wu, H.; Jin, J.; Zhang, W.; Grenz, A.; Eltzschig, H.K.; Tao, L.; et al. Elevated Endothelial Hypoxia-Inducible Factor-1alpha Contributes to Glomerular Injury and Promotes Hypertensive Chronic Kidney Disease. Hypertension 2015, 66, 75–84. [Google Scholar] [CrossRef]
- Eckardt, K.U.; Bernhardt, W.M.; Weidemann, A.; Warnecke, C.; Rosenberger, C.; Wiesener, M.S.; Willam, C. Role of hypoxia in the pathogenesis of renal disease. Kidney Int. Suppl. 2005, 99, S46–S51. [Google Scholar] [CrossRef]
- Kong, K.H.; Oh, H.J.; Lim, B.J.; Kim, M.; Han, K.H.; Choi, Y.H.; Kwon, K.; Nam, B.Y.; Park, K.S.; Park, J.T.; et al. Selective tubular activation of hypoxia-inducible factor-2alpha has dual effects on renal fibrosis. Sci. Rep. 2017, 7, 11351. [Google Scholar] [CrossRef]
- Schietke, R.E.; Hackenbeck, T.; Tran, M.; Gunther, R.; Klanke, B.; Warnecke, C.L.; Knaup, K.X.; Shukla, D.; Rosenberger, C.; Koesters, R.; et al. Renal tubular HIF-2alpha expression requires VHL inactivation and causes fibrosis and cysts. PLoS ONE 2012, 7, e31034. [Google Scholar] [CrossRef]
- Franco, M.; Bautista-Perez, R.; Perez-Mendez, O. Purinergic receptors in tubulointerstitial inflammatory cells: A pathophysiological mechanism of salt-sensitive hypertension. Acta Physiol. (Oxf) 2015, 214, 75–87. [Google Scholar] [CrossRef]
- Menzies, R.I.; Howarth, A.R.; Unwin, R.J.; Tam, F.W.; Mullins, J.J.; Bailey, M.A. Inhibition of the purinergic P2X7 receptor improves renal perfusion in angiotensin-II-infused rats. Kidney Int. 2015, 88, 1079–1087. [Google Scholar] [CrossRef]
- Maggiorani, D.; Dissard, R.; Belloy, M.; Saulnier-Blache, J.S.; Casemayou, A.; Ducasse, L.; Gres, S.; Belliere, J.; Caubet, C.; Bascands, J.L.; et al. Shear Stress-Induced Alteration of Epithelial Organization in Human Renal Tubular Cells. PLoS ONE 2015, 10, e0131416. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Z.; Gao, H.Q.; Liang, Y.; Zhang, J.; Wang, J.; Qiu, J. Cofilin1 is involved in hypertension-induced renal damage via the regulation of NF-kappaB in renal tubular epithelial cells. J. Transl. Med. 2015, 13, 323. [Google Scholar] [CrossRef]
- Hickson, L.J.; Eirin, A.; Lerman, L.O. Challenges and opportunities for stem cell therapy in patients with chronic kidney disease. Kidney Int. 2016, 89, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peired, A.J.; Sisti, A.; Romagnani, P. Mesenchymal Stem Cell-Based Therapy for Kidney Disease: A Review of Clinical Evidence. Stem Cells Int. 2016, 2016, 4798639. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sievers, L.K.; Eckardt, K.-U. Molecular Mechanisms of Kidney Injury and Repair in Arterial Hypertension. Int. J. Mol. Sci. 2019, 20, 2138. https://doi.org/10.3390/ijms20092138
Sievers LK, Eckardt K-U. Molecular Mechanisms of Kidney Injury and Repair in Arterial Hypertension. International Journal of Molecular Sciences. 2019; 20(9):2138. https://doi.org/10.3390/ijms20092138
Chicago/Turabian StyleSievers, Laura Katharina, and Kai-Uwe Eckardt. 2019. "Molecular Mechanisms of Kidney Injury and Repair in Arterial Hypertension" International Journal of Molecular Sciences 20, no. 9: 2138. https://doi.org/10.3390/ijms20092138
APA StyleSievers, L. K., & Eckardt, K. -U. (2019). Molecular Mechanisms of Kidney Injury and Repair in Arterial Hypertension. International Journal of Molecular Sciences, 20(9), 2138. https://doi.org/10.3390/ijms20092138