Novel Therapeutic Potentials of Taxifolin for Amyloid-β-associated Neurodegenerative Diseases and Other Diseases: Recent Advances and Future Perspectives

 ,
, 
Abstract
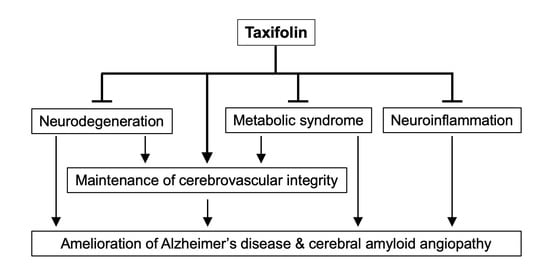
:
1. Introduction
2. Cerebral Amyloid Angiopathy
2.1. Overlapped Pathophysiologies Between Cerebral Amyloid Angiopathy and Alzheimer’s Disease
2.2. Strategies to Tackle Cerebral Amyloid Angiopathy
3. Therapeutic Potentials of Taxifolin for Cerebral Amyloid Angiopathy and Alzheimer’s Disease
3.1. Therapeutic Effects of Taxifolin on Cerebral Amyloid Angiopathy
3.2. Inhibitory Effects of Taxifolin on Amyloid-β42 Fibril Formation
3.3. Suppressive Effects of Taxifolin on Neuronal Amyloid-β Production
3.4. Potential Therapeutic Effects of Taxifolin on Alzheimer’s Disease
4. Therapeutic Potentials of Taxifolin for Metabolic Diseases with A High Risk for Neurodegenerative Diseases
4.1. Effects of Taxifolin on Diabetes
4.2. Effects of Taxifolin on Diabetic Nephropathy
4.3. Effects of Taxifolin on Obesity
5. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β |
| CAA | Cerebral amyloid angiopathy |
| IPAD | Intramural periarterial drainage |
References
- Rodrigues, M.A.; Samarasekera, N.; Lerpiniere, C.; Humphreys, C.; McCarron, M.O.; White, P.M.; Nicoll, J.A.R.; Sudlow, C.L.M.; Cordonnier, C.; Wardlaw, J.M.; et al. The Edinburgh CT and genetic diagnostic criteria for lobar intracerebral haemorrhage associated with cerebral amyloid angiopathy: Model development and diagnostic test accuracy study. Lancet Neurol. 2018, 17, 232–240. [Google Scholar] [CrossRef]
- Smith, E.E.; Greenberg, S.M. β-amyloid, blood vessels, and brain function. Stroke 2009, 40, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Sennfalt, S.; Norrving, B.; Petersson, J.; Ullberg, T. Long-Term Survival and Function After Stroke. Stroke 2018. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. Cerebral amyloid angiopathy: Emerging concepts. J. Stroke 2015, 17, 17–30. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Al-Shahi Salman, R.; Biessels, G.J.; van Buchem, M.; Cordonnier, C.; Lee, J.M.; Montaner, J.; Schneider, J.A.; Smith, E.E.; Vernooij, M.; et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol. 2014, 13, 419–428. [Google Scholar] [CrossRef]
- Charidimou, A.; Linn, J.; Vernooij, M.W.; Opherk, C.; Akoudad, S.; Baron, J.C.; Greenberg, S.M.; Jager, H.R.; Werring, D.J. Cortical superficial siderosis: Detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain 2015, 138, 2126–2139. [Google Scholar] [CrossRef]
- Chiang, G.C.; Cruz Hernandez, J.C.; Kantarci, K.; Jack, C.R.; Weiner, M.W.; Alzheimer’s Disease Neuroimaging Initiative. Cerebral Microbleeds, CSF p-Tau, and Cognitive Decline: Significance of Anatomic Distribution. AJNR Am. J. Neuroradiol. 2015, 36, 1635–1641. [Google Scholar] [CrossRef]
- Tsai, H.H.; Pasi, M.; Tsai, L.K.; Chen, Y.F.; Lee, B.C.; Tang, S.C.; Fotiadis, P.; Huang, C.Y.; Yen, R.F.; Gurol, M.E.; et al. Distribution of Lacunar Infarcts in Asians With Intracerebral Hemorrhage: A Magnetic Resonance Imaging and Amyloid Positron Emission Tomography Study. Stroke 2018, 49, 1515–1517. [Google Scholar] [CrossRef]
- Saito, S.; Yamamoto, Y.; Ihara, M. Mild Cognitive Impairment: At the Crossroad of Neurodegeneration and Vascular Dysfunction. Curr. Alzheimer Res. 2015, 12, 507–512. [Google Scholar] [CrossRef]
- Van Rooden, S.; Goos, J.D.; van Opstal, A.M.; Versluis, M.J.; Webb, A.G.; Blauw, G.J.; van der Flier, W.M.; Scheltens, P.; Barkhof, F.; van Buchem, M.A.; et al. Increased number of microinfarcts in Alzheimer disease at 7-T MR imaging. Radiology 2014, 270, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Westover, M.B.; Bianchi, M.T.; Yang, C.; Schneider, J.A.; Greenberg, S.M. Estimating cerebral microinfarct burden from autopsy samples. Neurology 2013, 80, 1365–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaria, R.N. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016, 131, 659–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, P.A.; Yu, L.; Nag, S.; Leurgans, S.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015, 85, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Evans, A.C.; Alzheimer’s Disease Neuroimaging, I. Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Comput. Biol. 2014, 10, e1003956. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Saito, S.; Ihara, M. New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 2014, 6, 290. [Google Scholar] [CrossRef]
- Nicoll, J.A.; Wilkinson, D.; Holmes, C.; Steart, P.; Markham, H.; Weller, R.O. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: A case report. Nat. Med. 2003, 9, 448–452. [Google Scholar] [CrossRef]
- Patton, R.L.; Kalback, W.M.; Esh, C.L.; Kokjohn, T.A.; Van Vickle, G.D.; Luehrs, D.C.; Kuo, Y.M.; Lopez, J.; Brune, D.; Ferrer, I.; et al. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: A biochemical analysis. Am. J. Pathol. 2006, 169, 1048–1063. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Kremer, B.P.; Rikkert, M.O.; Van Domburg, P.H.; Skehan, M.E.; Greenberg, S.M. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 2009, 66, 245–249. [Google Scholar] [CrossRef]
- van Etten, E.S.; Verbeek, M.M.; van der Grond, J.; Zielman, R.; van Rooden, S.; van Zwet, E.W.; van Opstal, A.M.; Haan, J.; Greenberg, S.M.; van Buchem, M.A.; et al. beta-Amyloid in CSF: Biomarker for preclinical cerebral amyloid angiopathy. Neurology 2017, 88, 169–176. [Google Scholar] [CrossRef]
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Mollgard, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018, 135, 363–385. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.W.J.; Carare, R.O.; Schreiber, S.; Hawkes, C.A. The Cerebrovascular Basement Membrane: Role in the Clearance of β-amyloid and Cerebral Amyloid Angiopathy. Front. Aging Neurosci. 2014, 6, 251. [Google Scholar] [CrossRef]
- Arbel-Ornath, M.; Hudry, E.; Eikermann-Haerter, K.; Hou, S.; Gregory, J.L.; Zhao, L.; Betensky, R.A.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 2013, 126, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.W.J.; Sharp, M.M.; Albargothy, N.J.; Fernandes, R.; Hawkes, C.A.; Verma, A.; Weller, R.O.; Carare, R.O. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016, 131, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.A.; Hartig, W.; Kacza, J.; Schliebs, R.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011, 121, 431–443. [Google Scholar] [CrossRef]
- Maki, T.; Okamoto, Y.; Carare, R.O.; Hase, Y.; Hattori, Y.; Hawkes, C.A.; Saito, S.; Yamamoto, Y.; Terasaki, Y.; Ishibashi-Ueda, H.; et al. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular β-amyloid. Ann. Clin. Transl. Neurol. 2014, 1, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Niwa, K.; Younkin, L.; Ebeling, C.; Turner, S.K.; Westaway, D.; Younkin, S.; Ashe, K.H.; Carlson, G.A.; Iadecola, C. Aβ1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. USA 2000, 97, 9735–9740. [Google Scholar] [CrossRef]
- Niwa, K.; Carlson, G.A.; Iadecola, C. Exogenous Aβ1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J. Cereb. Blood Flow Metab. 2000, 20, 1659–1668. [Google Scholar] [CrossRef]
- Park, L.; Zhou, P.; Koizumi, K.; El Jamal, S.; Previti, M.L.; Van Nostrand, W.E.; Carlson, G.; Iadecola, C. Brain and circulating levels of Aβ1-40 differentially contribute to vasomotor dysfunction in the mouse brain. Stroke 2013, 44, 198–204. [Google Scholar] [CrossRef]
- Weller, R.O.; Hawkes, C.A.; Carare, R.O.; Hardy, J. Does the difference between PART and Alzheimer’s disease lie in the age-related changes in cerebral arteries that trigger the accumulation of Abeta and propagation of tau? Acta Neuropathol. 2015, 129, 763–766. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Low-n oligomers as therapeutic targets of Alzheimer’s disease. J. Neurochem. 2011, 117, 19–28. [Google Scholar] [CrossRef]
- Ono, K.; Hamaguchi, T.; Naiki, H.; Yamada, M. Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochim. Biophys. Acta 2006, 1762, 575–586. [Google Scholar] [CrossRef]
- Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.; Masuda, Y.; Takegoshi, K.; Irie, K. Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 2013, 288, 23212–23224. [Google Scholar] [CrossRef]
- Hsu, F.; Park, G.; Guo, Z. Key Residues for the Formation of Abeta42 Amyloid Fibrils. ACS Omega 2018, 3, 8401–8407. [Google Scholar] [CrossRef]
- Park, L.; Koizumi, K.; El Jamal, S.; Zhou, P.; Previti, M.L.; Van Nostrand, W.E.; Carlson, G.; Iadecola, C. Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke 2014, 45, 1815–1821. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.L.; Abousaleh, F.; Brendza, R.P.; Dietrich, H.H.; Koenigsknecht-Talboo, J.; Cirrito, J.R.; Milner, E.; Holtzman, D.M.; Zipfel, G.J. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: Contribution of soluble and insoluble amyloid-β peptide, partial restoration via γ-secretase inhibition. J. Neurosci. 2008, 28, 13542–13550. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Youdim, K.A.; Shukitt-Hale, B.; Joseph, J.A. Flavonoids and the brain: Interactions at the blood-brain barrier and their physiological effects on the central nervous system. Free Radic. Biol. Med. 2004, 37, 1683–1693. [Google Scholar] [CrossRef]
- Yang, P.; Xu, F.; Li, H.F.; Wang, Y.; Li, F.C.; Shang, M.Y.; Liu, G.X.; Wang, X.; Cai, S.Q. Detection of 191 Taxifolin Metabolites and Their Distribution in Rats Using HPLC-ESI-IT-TOF-MSn. Molecules 2016, 21, 1209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Q.; Bao, X.; Ding, Y.; Shentu, J.; Cui, W.; Chen, X.; Wei, X.; Xu, S. Taxifolin prevents beta-amyloid-induced impairments of synaptic formation and deficits of memory via the inhibition of cytosolic phospholipase A2/prostaglandin E2 content. Metab. Brain Dis. 2018, 33, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.N.; Deeds, F. The toxicity and metabolism of dihydroquercetin. J. Am. Pharm. Assoc. Am. Pharm. Assoc. 1958, 47, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Schauss, A.G.; Tselyico, S.S.; Kuznetsova, V.A.; Yegorova, I. Toxicological and Genotoxicity Assessment of a Dihydroquercetin-Rich Dahurian Larch Tree (Larix gmelinii Rupr) Extract (Lavitol). Int. J. Toxicol. 2015, 34, 162–181. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Yamamoto, Y.; Maki, T.; Hattori, Y.; Ito, H.; Mizuno, K.; Harada-Shiba, M.; Kalaria, R.N.; Fukushima, M.; Takahashi, R.; et al. Taxifolin inhibits amyloid-beta oligomer formation and fully restores vascular integrity and memory in cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid β-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, H.Y.; Park, H.J.; Shin, H.K.; Hong, K.W.; Kim, C.D. Concurrent Treatment with Taxifolin and Cilostazol on the Lowering of beta-Amyloid Accumulation and Neurotoxicity via the Suppression of P-JAK2/P-STAT3/NF-kappaB/BACE1 Signaling Pathways. PLoS ONE 2016, 11, e0168286. [Google Scholar] [CrossRef]
- Sun, J.; Roy, S. The physical approximation of APP and BACE-1: A key event in Alzheimer’s disease pathogenesis. Dev. Neurobiol. 2018, 78, 340–347. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef]
- Stockley, J.H.; Ravid, R.; O’Neill, C. Altered beta-secretase enzyme kinetics and levels of both BACE1 and BACE2 in the Alzheimer’s disease brain. FEBS Lett. 2006, 580, 6550–6560. [Google Scholar] [CrossRef]
- Buggia-Prevot, V.; Sevalle, J.; Rossner, S.; Checler, F. NFkappaB-dependent control of BACE1 promoter transactivation by Abeta42. J. Biol. Chem. 2008, 283, 10037–10047. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell. Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef]
- Philippens, I.H.; Ormel, P.R.; Baarends, G.; Johansson, M.; Remarque, E.J.; Doverskog, M. Acceleration of Amyloidosis by Inflammation in the Amyloid-Beta Marmoset Monkey Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 101–113. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Hermann, P.M.; Watson, S.N.; Wildering, W.C. Phospholipase A2—nexus of aging, oxidative stress, neuronal excitability, and functional decline of the aging nervous system? Insights from a snail model system of neuronal aging and age-associated memory impairment. Front. Genet. 2014, 5, 419. [Google Scholar] [CrossRef]
- Sagy-Bross, C.; Kasianov, K.; Solomonov, Y.; Braiman, A.; Friedman, A.; Hadad, N.; Levy, R. The role of cytosolic phospholipase A2 alpha in amyloid precursor protein induction by amyloid beta1-42 : Implication for neurodegeneration. J. Neurochem. 2015, 132, 559–571. [Google Scholar] [CrossRef]
- Qu, B.; Gong, Y.; Gill, J.M.; Kenney, K.; Diaz-Arrastia, R. Heterozygous knockout of cytosolic phospholipase A2alpha attenuates Alzheimer’s disease pathology in APP/PS1 transgenic mice. Brain Res. 2017, 1670, 248–252. [Google Scholar] [CrossRef]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef]
- Bate, C.; Williams, A. cAMP-Inhibits Cytoplasmic Phospholipase A(2) and Protects Neurons against Amyloid-beta-Induced Synapse Damage. Biology (Basel) 2015, 4, 591–606. [Google Scholar] [PubMed]
- Brummett, A.M.; Navratil, A.R.; Bryan, J.D.; Woolard, M.D. Janus kinase 3 activity is necessary for phosphorylation of cytosolic phospholipase A2 and prostaglandin E2 synthesis by macrophages infected with Francisella tularensis live vaccine strain. Infect. Immun. 2014, 82, 970–982. [Google Scholar] [CrossRef]
- Bate, C.; Williams, A. Alpha-Synuclein-induced synapse damage in cultured neurons is mediated by cholesterol-sensitive activation of cytoplasmic phospholipase A2. Biomolecules 2015, 5, 178–193. [Google Scholar] [CrossRef]
- Ohara, T.; Doi, Y.; Ninomiya, T.; Hirakawa, Y.; Hata, J.; Iwaki, T.; Kanba, S.; Kiyohara, Y. Glucose tolerance status and risk of dementia in the community: The Hisayama study. Neurology 2011, 77, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Peters, S.A.; Woodward, M.; Mejia Arango, S.; Batty, G.D.; Beckett, N.; Beiser, A.; Borenstein, A.R.; Crane, P.K.; Haan, M.; et al. Type 2 Diabetes as a Risk Factor for Dementia in Women Compared With Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes Care 2016, 39, 300–307. [Google Scholar] [CrossRef]
- Morley, J.E. Diabetes: The diabetic brain. Nat. Rev. Endocrinol. 2017, 13, 570–571. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Hirabayashi, N.; Hata, J.; Ohara, T.; Mukai, N.; Nagata, M.; Shibata, M.; Gotoh, S.; Furuta, Y.; Yamashita, F.; Yoshihara, K.; et al. Association Between Diabetes and Hippocampal Atrophy in Elderly Japanese: The Hisayama Study. Diabetes Care 2016, 39, 1543–1549. [Google Scholar] [CrossRef] [Green Version]
- Rehman, K.; Chohan, T.A.; Waheed, I.; Gilani, Z.; Akash, M.S.H. Taxifolin prevents postprandial hyperglycemia by regulating the activity of alpha-amylase: Evidence from an in vivo and in silico studies. J. Cell. Biochem. 2019, 120, 425–438. [Google Scholar] [CrossRef]
- Rosak, C.; Mertes, G. Critical evaluation of the role of acarbose in the treatment of diabetes: Patient considerations. Diabetes Metab. Syndr. Obes. 2012, 5, 357–367. [Google Scholar] [CrossRef]
- Ding, T.; Wang, S.; Zhang, X.; Zai, W.; Fan, J.; Chen, W.; Bian, Q.; Luan, J.; Shen, Y.; Zhang, Y.; et al. Kidney protection effects of dihydroquercetin on diabetic nephropathy through suppressing ROS and NLRP3 inflammasome. Phytomedicine 2018, 41, 45–53. [Google Scholar] [CrossRef]
- Seliger, S.L.; Siscovick, D.S.; Stehman-Breen, C.O.; Gillen, D.L.; Fitzpatrick, A.; Bleyer, A.; Kuller, L.H. Moderate renal impairment and risk of dementia among older adults: The Cardiovascular Health Cognition Study. J. Am. Soc. Nephrol. 2004, 15, 1904–1911. [Google Scholar] [CrossRef]
- Helmer, C.; Stengel, B.; Metzger, M.; Froissart, M.; Massy, Z.A.; Tzourio, C.; Berr, C.; Dartigues, J.F. Chronic kidney disease, cognitive decline, and incident dementia: The 3C Study. Neurology 2011, 77, 2043–2051. [Google Scholar] [CrossRef]
- Takae, K.; Hata, J.; Ohara, T.; Yoshida, D.; Shibata, M.; Mukai, N.; Hirakawa, Y.; Kishimoto, H.; Tsuruya, K.; Kitazono, T.; et al. Albuminuria Increases the Risks for Both Alzheimer Disease and Vascular Dementia in Community-Dwelling Japanese Elderly: The Hisayama Study. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Fujisaki, K.; Tsuruya, K.; Yamato, M.; Toyonaga, J.; Noguchi, H.; Nakano, T.; Taniguchi, M.; Tokumoto, M.; Hirakata, H.; Kitazono, T. Cerebral oxidative stress induces spatial working memory dysfunction in uremic mice: Neuroprotective effect of tempol. Nephrol. Dial. Transplant. 2014, 29, 529–538. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, W.; Wang, J.; Chen, Y.; Huang, W.; Zhu, Y. Taxifolin attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Am. J. Transl. Res. 2018, 10, 1205–1210. [Google Scholar]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef]
- Qizilbash, N.; Gregson, J.; Johnson, M.E.; Pearce, N.; Douglas, I.; Wing, K.; Evans, S.J.W.; Pocock, S.J. BMI and risk of dementia in two million people over two decades: A retrospective cohort study. Lancet Diabetes Endocrinol. 2015, 3, 431–436. [Google Scholar] [CrossRef]
- Collaboration, N.C.D.R.F. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar]
- Moll van Charante, E.P.; Richard, E.; Eurelings, L.S.; van Dalen, J.W.; Ligthart, S.A.; van Bussel, E.F.; Hoevenaar-Blom, M.P.; Vermeulen, M.; van Gool, W.A. Effectiveness of a 6-year multidomain vascular care intervention to prevent dementia (preDIVA): A cluster-randomised controlled trial. Lancet 2016, 388, 797–805. [Google Scholar] [CrossRef]
- Zekry, S.H.; Abo-elmatty, D.M.; Zayed, R.A.; Radwan, M.M.; ElSohly, M.A.; Hassanean, H.A.; Ahmed, S.A. Effect of metabolites isolated from Cuscuta pedicellata on high fat diet-fed rats. Med. Chem. Res. 2015, 24, 1964–1973. [Google Scholar] [CrossRef]
- Mehanna, E.T.; El-Sayed, N.M.; Ibrahim, A.K.; Ahmed, S.A.; Abo-Elmatty, D.M. Isolated compounds from Cuscuta pedicellata ameliorate oxidative stress and upregulate expression of some energy regulatory genes in high fat diet induced obesity in rats. Biomed. Pharmacother. 2018, 108, 1253–1258. [Google Scholar] [CrossRef]
- Saito, S.; Kojima, S.; Oishi, N.; Kakuta, R.; Maki, T.; Yasuno, F.; Nagatsuka, K.; Yamamoto, H.; Fukuyama, H.; Fukushima, M.; et al. A multicenter, randomized, placebo-controlled trial for cilostazol in patients with mild cognitive impairment: The COMCID study protocol. Alzheimers Dement (NY) 2016, 2, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Inoue, T.; Saito, S.; Tanaka, M.; Yamakage, H.; Kusakabe, T.; Shimatsu, A.; Ihara, M.; Satoh-Asahara, N. Pleiotropic neuroprotective effects of taxifolin in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Pharmacological Effects | Targets | Mechanisms |
|---|---|---|
| Suppressing Aβ production | Neuron | Reduction of BACE1 levels |
| Inhibiting Aβ aggregation | Lys residues of Aβ | Aβ–taxifolin adduct formation |
| Anti-inflammation | Neuron | Reduction of cPLA2 and PGE2 levels |
| Increasing CBF and CVR | Vascular endothelial and/or mural cells | Amelioration of Aβ toxicity Anti-oxidation Anti-glycation |
| Reducing hyperglycemia | α-amylase | Taxifolin–α-amylase complex |
| Reducing body weight | Brown adipose tissue | Increased energy expenditure |
| Renal protective effects in diabetic conditions | Renal tissue | Anti-fibrosis Anti-oxidation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, M.; Saito, S.; Inoue, T.; Satoh-Asahara, N.; Ihara, M. Novel Therapeutic Potentials of Taxifolin for Amyloid-β-associated Neurodegenerative Diseases and Other Diseases: Recent Advances and Future Perspectives. Int. J. Mol. Sci. 2019, 20, 2139. https://doi.org/10.3390/ijms20092139
Tanaka M, Saito S, Inoue T, Satoh-Asahara N, Ihara M. Novel Therapeutic Potentials of Taxifolin for Amyloid-β-associated Neurodegenerative Diseases and Other Diseases: Recent Advances and Future Perspectives. International Journal of Molecular Sciences. 2019; 20(9):2139. https://doi.org/10.3390/ijms20092139
Chicago/Turabian StyleTanaka, Masashi, Satoshi Saito, Takayuki Inoue, Noriko Satoh-Asahara, and Masafumi Ihara. 2019. "Novel Therapeutic Potentials of Taxifolin for Amyloid-β-associated Neurodegenerative Diseases and Other Diseases: Recent Advances and Future Perspectives" International Journal of Molecular Sciences 20, no. 9: 2139. https://doi.org/10.3390/ijms20092139
APA StyleTanaka, M., Saito, S., Inoue, T., Satoh-Asahara, N., & Ihara, M. (2019). Novel Therapeutic Potentials of Taxifolin for Amyloid-β-associated Neurodegenerative Diseases and Other Diseases: Recent Advances and Future Perspectives. International Journal of Molecular Sciences, 20(9), 2139. https://doi.org/10.3390/ijms20092139