ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy

Abstract
:
1. Introduction
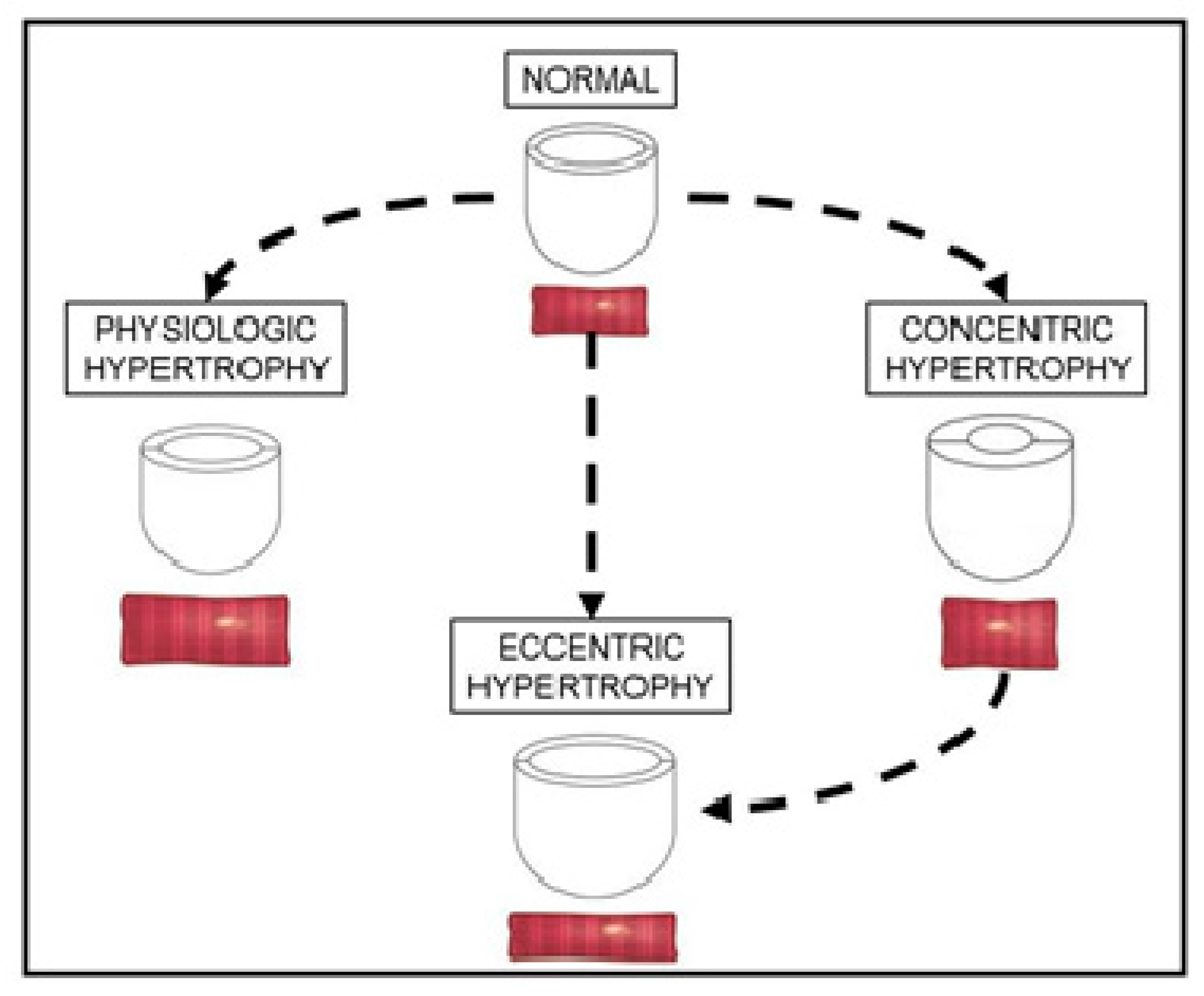
2. Overview of Cardiac Hypertrophy
3. The Role of ERK in Adaptive Cardiac Hypertrophy
3.1. Adaptive Concentric Hypertrophy
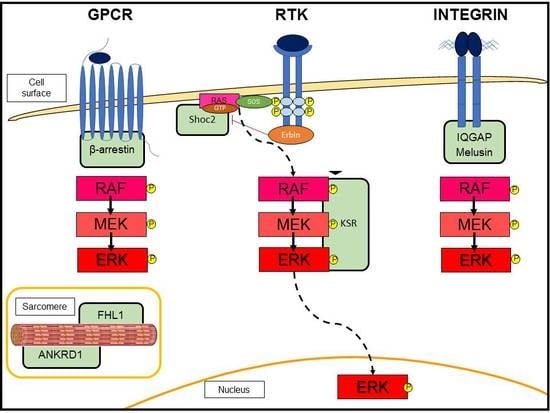
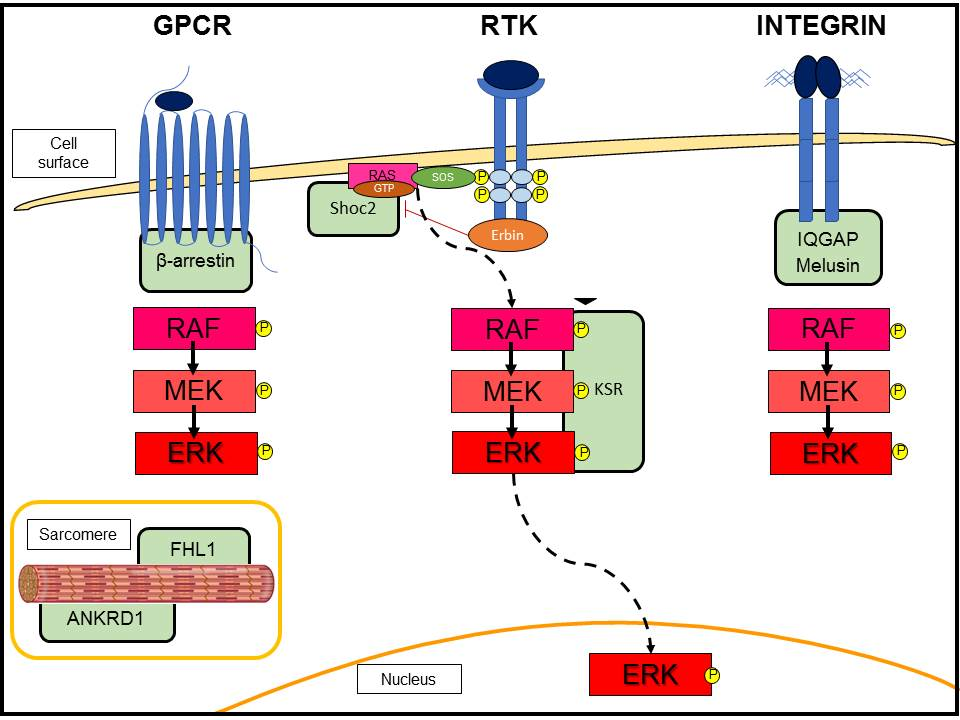
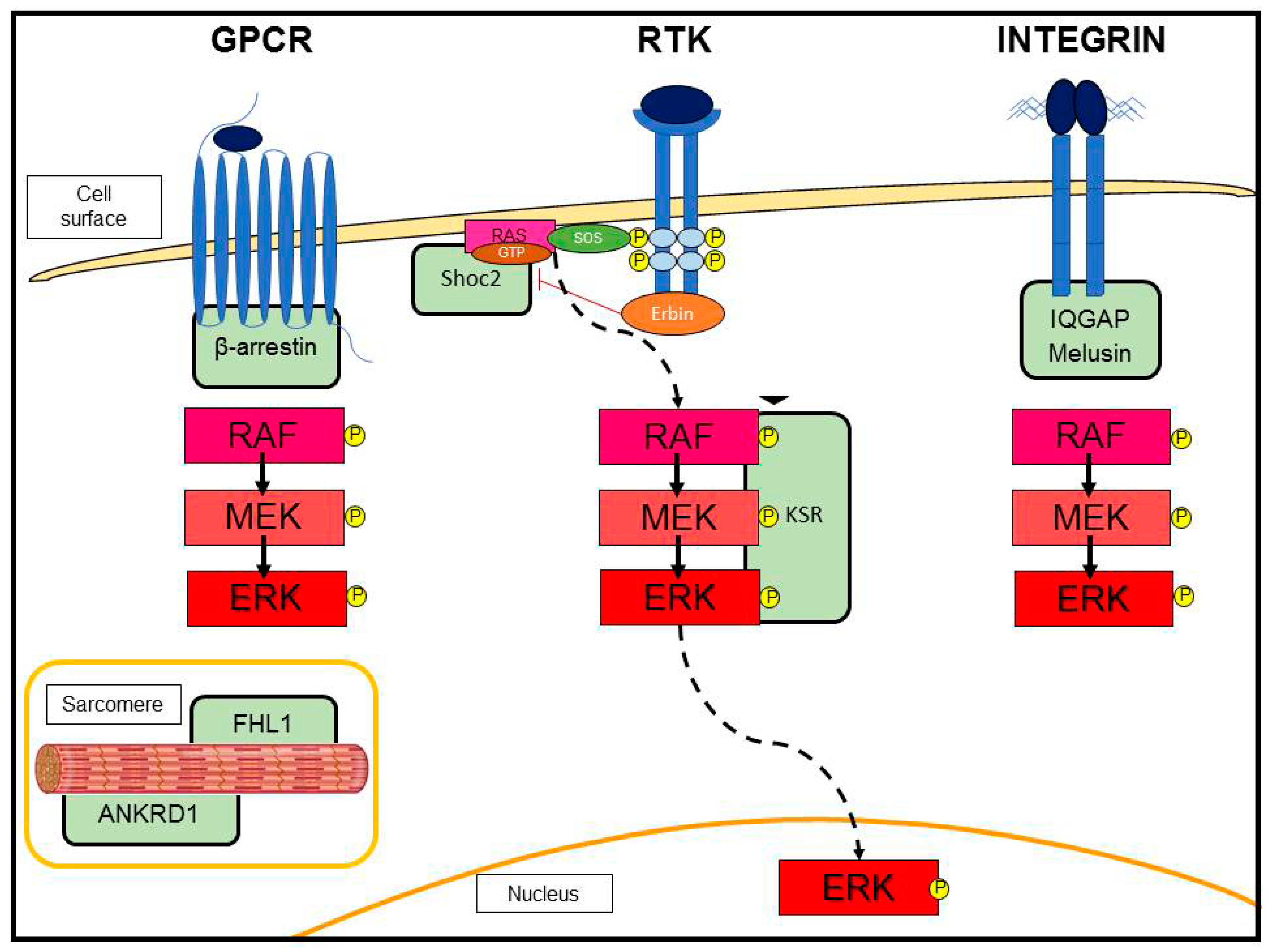
3.2. G-Protein-Coupled Receptor (GPCR)-Induced Adaptive Hypertrophy
3.3. Scaffold Proteins in ERK Signaling
3.4. Cell Death Prevention
4. The Role of ERK in Maladaptive Cardiac Hypertrophy
4.1. Hypertension
4.2. Anthracycline-Induced Cardiotoxicity (CTX)
4.3. ERK Phosphorylation at Threonine 188 (T188)
4.4. ERK5
5. ERK Activity in Mice Overexpressing RTKs in Cardiomyocytes
6. ERK in Genetic Diseases with Hypertrophy
6.1. Hypertrophic Cardiomyopathies (HCMs)
6.2. RASopathies
7. Targeting ERK to Therapeutically Modulate the Cardiac Hypertrophy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| αAR | α-adrenergic receptor |
| αTM | α tropomyosin |
| AngII | angiotensin II |
| ANKRD1 | ankyrin repeat domain 1 |
| βAR | β-adrenergic receptor |
| βMHC | β myosin heavy chain |
| BMK1 | big MAPK 1 |
| CTX | cardiotoxicity |
| cTnC | cardiac troponin C |
| cTnI | cardiac troponin I |
| cTnT | cardiac troponin T |
| Doxo | doxorubicin |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| ErbB2 | erythroblastic leukemia viral oncogene homolog 2 |
| ERK | extracellular signal-regulated kinase |
| FGF | fibroblast growth factor |
| FHL1 | four and a half LIM domain protein-1 |
| GPCR | G-protein-coupled receptor |
| GSK3 | glycogen synthase kinase III |
| HCM | hypertrophic cardiomyopathy |
| HER2 | epidermal growth factor receptor 2 |
| HGFR | hepatocyte growth factor receptor |
| HSF | heat shock factor |
| IGFR | insulin-like growth factor receptor |
| IQGAP1 | IQ motif-containing GTPase-activating protein 1 |
| LAMP-2 | lysosome-associated membrane protein 2 |
| MAPK | mitogen-activated protein kinase |
| MI | myocardial infarction |
| NF-κB | nuclear factor kappa B |
| PRKAG2 | γ-2-regulatory subunit of the AMP-activated protein kinase |
| PTPN11 | protein tyrosine phosphatase, nonreceptor type 11 |
| ROS | reactive oxygen species |
| RTK | receptor tyrosine kinase |
| TAC | transverse aortic constriction |
| T188 | threonine 188 |
| Tg | transgenic mice |
References
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of a Three-Kinase Module from Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Gerits, N.; Kostenko, S.; Moens, U. In vivo functions of mitogen-activated protein kinases: conclusions from knock-in and knock-out mice. Transgenic. Res. 2007, 16, 281–314. [Google Scholar] [CrossRef]
- Bueno, O.F.; De Windt, L.J.; Tymitz, K.M.; Witt, S.A.; Kimball, T.R.; Klevitsky, R.; Hewett, T.E.; Jones, S.P.; Lefer, D.J.; Peng, C.F.; et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000, 19, 6341–6350. [Google Scholar] [CrossRef]
- Clerk, A.; Sugden, P.H. Signaling through the extracellular signal-regulated kinase 1/2 cascade in cardiac myocytes. Biochem. Cell. Biol. 2004, 82, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Petrich, B.G.; Wang, Y. Stress-Activated MAP Kinases in Cardiac Remodeling and Heart Failure: New Insights from Transgenic Studies. Trends Cardiovasc. Med. 2004, 14, 50–55. [Google Scholar] [CrossRef]
- Wang, Y. Mitogen-activated protein kinases in heart development and diseases. Circulation 2007, 116, 1413–1423. [Google Scholar] [CrossRef]
- Sala, V.; Gallo, S.; Leo, C.; Gatti, S.; Gelb, B.D.; Crepaldi, T. Signaling to cardiac hypertrophy: Insights from human and mouse RASopathies. Mol. Med. 2012, 18, 938–947. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular Pathways Underlying Cardiac Remodeling During Pathophysiological Stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Nicol, R.L.; Frey, N.; Pearson, G.; Cobb, M.; Richardson, J.; Olson, E.N. Activated MEK5 induces serial assembly of sarcomeres and eccentric cardiac hypertrophy. EMBO J. 2001, 20, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Cardiac Hypertrophy: The Good, the Bad, and the Ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef]
- MacLellan, W.R.; Schneider, M.D. Genetic Dissection of Cardiac Growth Control Pathways. Annu. Rev. Physiol. 2000, 62, 289–320. [Google Scholar] [CrossRef]
- Selvetella, G.; Hirsch, E.; Notte, A.; Tarone, G.; Lembo, G. Adaptive and maladaptive hypertrophic pathways: points of convergence and divergence. Cardiovasc. Res. 2004, 63, 373–380. [Google Scholar] [CrossRef]
- Vakili, B.A.; Okin, P.M.; Devereux, R.B. Prognostic implications of left ventricular hypertrophy. Am. Heart. J. 2001, 141, 334–341. [Google Scholar] [CrossRef]
- Maillet, M.; Van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell. Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Wachhorst, S.P.; Mao, L.; Ross, J. ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am. J. Physiol. 1994, 266, H2468–H2475. [Google Scholar] [CrossRef]
- Dorn, G.W.; Robbins, J.; Ball, N.; Walsh, R.A. Myosin heavy chain regulation and myocyte contractile depression after LV hypertrophy in aortic-banded mice. Am. J. Physiol. 1994, 267, H400–H405. [Google Scholar] [CrossRef]
- Li, X.M.; Ma, Y.T.; Yang, Y.N.; Liu, F.; Chen, B.D.; Han, W.; Zhang, J.F.; Gao, X.M. Downregulation of survival signalling pathways and increased apoptosis in the transition of pressure overload-induced cardiac hypertrophy to heart failure. Clin. Exp. Pharmacol. Physiol. 2009, 36, 1054–1061. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2000, 98, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Wei, H.; Garrison, T.R.; Lefkowitz, R.J. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by beta-arrestins 1 and 2. J. Biol. Chem. 2004, 279, 7807–7811. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of Receptor Signals by beta-Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Reiter, E.; Premont, R.T.; Lichtarge, O.; Lefkowitz, R.J. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 2006, 281, 1261–1273. [Google Scholar] [CrossRef]
- Salazar, N.C.; Chen, J.; Rockman, H.A. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 2007, 1768, 1006–1018. [Google Scholar] [CrossRef]
- Brancaccio, M.; Hirsch, E.; Notte, A.; Selvetella, G.; Lembo, G.; Tarone, G. Integrin signalling: The tug-of-war in heart hypertrophy. Cardiovasc. Res. 2006, 70, 422–433. [Google Scholar] [CrossRef]
- Tavi, P.; Laine, M.; Weckström, M.; Ruskoaho, H. Cardiac mechanotransduction: from sensing to disease and treatment. Trends Pharmacol. Sci. 2001, 22, 254–260. [Google Scholar] [CrossRef]
- Miller, W.E.; Lefkowitz, R.J. Expanding roles for β-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr. Opin. Cell. Biol. 2001, 13, 139–145. [Google Scholar] [CrossRef]
- Noma, T.; Lemaire, A.; Naga Prasad, S.V.; Barki-Harrington, L.; Tilley, D.G.; Chen, J.; Le Corvoisier, P.; Violin, J.D.; Wei, H.; Lefkowitz, R.J.; et al. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Investig. 2007, 117, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.M.; Tilley, D.G.; Chen, J.; Salazar, N.C.; Whalen, E.J.; Violin, J.D.; Rockman, H.A. Blockers alprenolol and carvedilol stimulate-arrestin-mediated EGFR transactivation. Proc. Natl. Acad. Sci. USA 2008, 105, 14555–14560. [Google Scholar] [CrossRef]
- Harris, I.S.; Zhang, S.; Treskov, I.; Kovacs, A.; Weinheimer, C.; Muslin, A.J. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation 2004, 110, 718–723. [Google Scholar] [CrossRef]
- Mutlak, M.; Schlesinger-Laufer, M.; Haas, T.; Shofti, R.; Ballan, N.; Lewis, Y.E.; Zuler, M.; Zohar, Y.; Caspi, L.H.; Kehat, I. Extracellular signal-regulated kinase (ERK) activation preserves cardiac function in pressure overload induced hypertrophy. Int. J. Cardiol. 2018, 270, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.D.; Ishizaka, S.; Nakamura, A.; Swigart, P.M.; Rodrigo, M.C.; Simpson, G.L.; Cotecchia, S.; Rokosh, D.G.; Grossman, W.; Foster, E.; et al. The alpha(1A/C)- and alpha(1B)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J. Clin. Investig. 2003, 111, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.C.; Fan, Y.; Xu, Y.; Yang, Y.L.; Simpson, P.C.; Mann, M.J. Shift toward greater pathologic post-myocardial infarction remodeling with loss of the adaptive hypertrophic signaling of alpha1 adrenergic receptors in mice. PLoS ONE 2017, 12, e0188471. [Google Scholar] [CrossRef]
- Patel, P.A.; Tilley, D.G.; Rockman, H.A. Beta-arrestin-mediated signaling in the heart. Circ. J. 2008, 72, 1725–1729. [Google Scholar] [CrossRef]
- Morrison, D.K.; Davis, R.J. Regulation of MAP Kinase Signaling Modules by Scaffold Proteins in Mammals. Annu. Rev. Cell. Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef]
- Kornfeld, K.; Hom, D.B.; Horvitz, H.R. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 1995, 83, 903–913. [Google Scholar] [CrossRef]
- Brown, M.D.; Sacks, D.B. Protein scaffolds in MAP kinase signalling. Cell Signal 2009, 21, 462–469. [Google Scholar] [CrossRef]
- Liang, Y.; Sheikh, F. Scaffold proteins regulating extracellular regulated kinase function in cardiac hypertrophy and disease. Front. Pharmacol. 2016, 7, 37. [Google Scholar] [CrossRef]
- Shi, M.; Zhao, M.; Hu, M.; Liu, D.; Cao, H.; Qian, L.; Yang, Z.; Hu, Y.; Yu, M.; Yang, S.; et al. β2-AR-induced Her2 transactivation mediated by Erbin confers protection from apoptosis in cardiomyocytes. Int. J. Cardiol. 2013, 167, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Rachmin, I.; Tshori, S.; Smith, Y.; Oppenheim, A.; Marchetto, S.; Kay, G.; Foo, R.S.; Dagan, N.; Golomb, E.; Gilon, D.; et al. Erbin is a negative modulator of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 5902–5907. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.R.; Galperin, E. The function of Shoc2: A scaffold and beyond. Commun. Integr. Biol. 2016, 9, e1188241. [Google Scholar] [CrossRef] [PubMed]
- Sbroggiò, M.; Carnevale, D.; Bertero, A.; Cifelli, G.; De Blasio, E.; Mascio, G.; Hirsch, E.; Bahou, W.F.; Turco, E.; Silengo, L.; et al. IQGAP1 regulates ERK1/2 and AKT signalling in the heart and sustains functional remodelling upon pressure overload. Cardiovasc. Res. 2011, 91, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, R.; Sbroggiò, M.; Di Savino, A.; Fusella, F.; Bertero, A.; Michowski, W.; Tarone, G.; Brancaccio, M. Morgana and melusin: Two fairies chaperoning signal transduction. Cell Cycle 2011, 10, 3678–3683. [Google Scholar] [CrossRef]
- Brancaccio, M.; Fratta, L.; Notte, A.; Hirsch, E.; Poulet, R.; Guazzone, S.; De Acetis, M.; Vecchione, C.; Marino, G.; Altruda, F.; et al. Melusin, a muscle-specific integrin β1–interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 2003, 9, 68–75. [Google Scholar] [CrossRef]
- De Acetis, M.; Notte, A.; Accornero, F.; Selvetella, G.; Brancaccio, M.; Vecchione, C.; Sbroggiò, M.; Collino, F.; Pacchioni, B.; Lanfranchi, G.; et al. Cardiac Overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circ. Res. 2005, 96, 1087–1094. [Google Scholar] [CrossRef]
- Sheikh, F.; Raskin, A.; Chu, P.H.; Lange, S.; Domenighetti, A.A.; Zheng, M.; Liang, X.; Zhang, T.; Yajima, T.; Gu, Y.; et al. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Investig. 2008, 118, 3870–3880. [Google Scholar] [CrossRef]
- Bang, M.L.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Chien, K.R.; Chen, J. The muscle ankyrin repeat proteins CARP, Ankrd2, and DARP are not essential for normal cardiac development and function at basal conditions and in response to pressure overload. PLoS ONE 2014, 9, e93638. [Google Scholar] [CrossRef]
- Maeda, T.; Sepulveda, J.; Chen, H.H.; Stewart, A.F.R. α1-Adrenergic activation of the cardiac ankyrin repeat protein gene in cardiac myocytes. Gene 2002, 297, 1–9. [Google Scholar] [CrossRef]
- Zhong, L.; Chiusa, M.; Cadar, A.G.; Lin, A.; Samaras, S.; Davidson, J.M.; Lim, C.C. Targeted inhibition of ANKRD1 disrupts sarcomeric ERK-GATA4 signal transduction and abrogates phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc. Res. 2015, 106, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Cai, H.; Zhao, Z.M.; Chang, W.J.; Gu, N.; Cao, S.P.; Wu, M.L. Icariin attenuated oxidative stress induced-cardiac apoptosis by mitochondria protection and ERK activation. Biomed. Pharmacother. 2016, 83, 1089–1094. [Google Scholar] [CrossRef]
- Liang, Q.; Wiese, R.J.; Bueno, O.F.; Dai, Y.S.; Markham, B.E.; Molkentin, J.D. The transcription factor GATA4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol. Cell. Biol. 2001, 21, 7460–7469. [Google Scholar] [CrossRef] [PubMed]
- Aries, A.; Paradis, P.; Lefebvre, C.; Schwartz, R.J.; Nemer, M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 6975–6980. [Google Scholar] [CrossRef]
- Tran, S.E.; Holmstrom, T.H.; Ahonen, M.; Kahari, V.M.; Eriksson, J.E. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J. Biol. Chem. 2001, 276, 16484–16490. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, J.; Ohmichi, M.; Kurachi, H.; Kanda, Y.; Hisamoto, K.; Nishio, Y.; Adachi, K.; Tasaka, K.; Kanzaki, T.; Murata, Y. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res. 2000, 60, 5988–5994. [Google Scholar] [PubMed]
- Biswas, S.C.; Greene, L.A. Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J. Biol. Chem. 2002, 277, 49511–49516. [Google Scholar] [CrossRef]
- Allan, L.A.; Morrice, N.; Brady, S.; Magee, G.; Pathak, S.; Clarke, P.R. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat. Cell. Biol. 2003, 5, 647–654. [Google Scholar] [CrossRef]
- Domina, A.M.; Vrana, J.A.; Gregory, M.A.; Hann, S.R.; Craig, R.W. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 2004, 23, 5301–5315. [Google Scholar] [CrossRef]
- Garcia, J.; Ye, Y.; Arranz, V.; Letourneux, C.; Pezeron, G.; Porteu, F. IEX-1: a new ERK substrate involved in both ERK survival activity and ERK activation. EMBO J. 2002, 21, 5151–5163. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Kim, S.; Ohta, K.; Hamaguchi, A.; Yunico, T.; Miura, K.; Iwao, H. Angiotensin II induces cardiac phenotypic modulation and remodeling in vivo in rats. Hypertens 1995, 25, 1252–1259. [Google Scholar] [CrossRef]
- Suzuki, J.; Matsubara, H.; Urakami, M.; Inada, M. Rat angiotensin II (type 1A) receptor mRNA regulation and subtype expression in myocardial growth and hypertrophy. Circ. Res. 1993, 73, 439–447. [Google Scholar] [CrossRef]
- Paradis, P.; Dali-Youcef, N.; Paradis, F.W.; Thibault, G.; Nemer, M. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 931–936. [Google Scholar] [CrossRef]
- Harada, K.; Sugaya, T.; Murakami, K.; Yazaki, Y.; Komuro, I. Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation 1999, 100, 2093–2099. [Google Scholar] [CrossRef]
- Pellieux, C.; Sauthier, T.; Aubert, J.F.; Brunner, H.R.; Pedrazzini, T. Angiotensin II-induced cardiac hypertrophy is associated with different mitogen-activated protein kinase activation in normotensive and hypertensive mice. J. Hypertens. 2000, 18, 1307–1317. [Google Scholar] [CrossRef]
- Yamazaki, T.; Komuro, I.; Yazaki, Y. Role of the renin-angiotensin system in cardiac hypertrophy. Am. J. Cardiol. 1999, 83, 53–57. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lee, F.L.; Peng, S.F.; Lin, K.H.; Chen, R.J.; Ho, T.J.; Tsai, F.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. HSF1 phosphorylation by ERK/GSK3 suppresses RNF126 to sustain IGF-IIR expression for hypertension-induced cardiomyocyte hypertrophy. J. Cell. Physiol. 2018, 233, 979–989. [Google Scholar] [CrossRef]
- Zhai, P.; Yamamoto, M.; Galeotti, J.; Liu, J.; Masurekar, M.; Thaisz, J.; Irie, K.; Holle, E.; Yu, X.; Kupershmidt, S.; et al. Cardiac-specific overexpression of AT1 receptor mutant lacking G alpha q/G alpha i coupling causes hypertrophy and bradycardia in transgenic mice. J. Clin. Investig. 2005, 115, 3045–3056. [Google Scholar] [CrossRef]
- Chen, Q.M.; Tu, V.C.; Purdon, S.; Wood, J.; Dilley, T. Molecular mechanisms of cardiac hypertrophy induced by toxicants. Cardiovasc. Toxicol. 2001, 1, 267–283. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chen, J.Y.; Kuo, C.H.; Pai, P.Y.; Ho, T.J.; Chen, T.S.; Tsai, F.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. Mitochondrial ROS-induced ERK1/2 activation and HSF2-mediated AT1R upregulation are required for doxorubicin-induced cardiotoxicity. J. Cell. Physiol. 2018, 233, 463–475. [Google Scholar] [CrossRef]
- Liu, M.H.; Lin, X.L.; Zhang, Y.; He, J.; Tan, T.P.; Wu, S.J.; Liu, J.; Tian, W.; Chen, L.; Yu, S.; et al. Hydrogen sulfide attenuates doxorubicin-induced cardiotoxicity by inhibiting reactive oxygen species-activated extracellular signal-regulated kinase 1/2 in H9c2 cardiac myocytes. Mol. Med. Rep. 2015, 12, 6841–6848. [Google Scholar] [CrossRef]
- Tang, F.; Zhou, X.; Wang, L.; Shan, L.; Li, C.; Zhou, H.; Lee, S.M.; Hoi, M.P. A novel compound DT-010 protects against doxorubicin-induced cardiotoxicity in zebrafish and H9c2 cells by inhibiting reactive oxygen species-mediated apoptotic and autophagic pathways. Eur. J. Pharmacol. 2018, 820, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Loh, S.H.; Chen, J.J.; Tsai, C.S. Urotensin II prevents cardiomyocyte apoptosis induced by doxorubicin via Akt and ERK. Eur. J. Pharmacol. 2012, 680, 88–94. [Google Scholar] [CrossRef] [PubMed]
- He, S.F.; Jin, S.Y.; Wu, H.; Wang, B.; Wu, Y.X.; Zhang, S.J.; Irwin, M.G.; Wong, T.M.; Zhang, Y. Morphine preconditioning confers cardioprotection in doxorubicin-induced failing rat hearts via ERK/GSK-3β pathway independent of PI3K/Akt. Toxicol. Appl. Pharmacol. 2015, 288, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Danelisen, I.; Singal, P.K. Involvement of mitogen-activated protein kinases in adriamycin-induced cardiomyopathy. Am. J. Physiol. Circ. Physiol. 2005, 288, H1925–H1930. [Google Scholar] [CrossRef]
- Nemeth, B.T.; Varga, Z.V.; Wu, W.J.; Pacher, P. Trastuzumab cardiotoxicity: from clinical trials to experimental studies. Br. J. Pharmacol. 2017, 174, 3727–3748. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef]
- Milano, G.; Raucci, A.; Scopece, A.; Daniele, R.; Guerrini, U.; Sironi, L.; Cardinale, D.; Capogrossi, M.C.; Pompilio, G. Doxorubicin and trastuzumab regimen induces biventricular failure in mice. J. Am. Soc. Echocardiogr. 2014, 27, 568–579. [Google Scholar] [CrossRef]
- Crone, S.A.; Zhao, Y.Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar] [CrossRef]
- Fukazawa, R.; Miller, T.A.; Kuramochi, Y.; Frantz, S.; Kim, Y.D.; Marchionni, M.A.; Kelly, R.A.; Sawyer, D.B. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J. Mol. Cell. Cardiol. 2003, 35, 1473–1479. [Google Scholar] [CrossRef]
- Rochette, L.; Guenancia, C.; Gudjoncik, A.; Hachet, O.; Zeller, M.; Cottin, Y.; Vergely, C. Anthracyclines/trastuzumab: new aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol. Sci. 2015, 36, 326–348. [Google Scholar] [CrossRef]
- Negro, A.; Brar, B.K.; Lee, K.F. Essential roles of Her2/erbB2 in cardiac development and function. Recent. Prog. Horm. Res. 2004, 59, 1–12. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Patel, D.; Wu, X. The dual-targeted HER1/HER2 tyrosine kinase inhibitor lapatinib strongly potentiates the cardiac myocyte-damaging effects of doxorubicin. Cardiovasc. Toxicol. 2013, 13, 33–47. [Google Scholar] [CrossRef]
- Mohan, N.; Shen, Y.; Endo, Y.; ElZarrad, M.K.; Wu, W.J. Trastuzumab, but Not Pertuzumab, Dysregulates HER2 Signaling to Mediate Inhibition of Autophagy and Increase in Reactive Oxygen Species Production in Human Cardiomyocytes. Mol. Cancer Ther. 2016, 15, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Schmitt, J.P.; Schmitteckert, E.M.; Lohse, M.J. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 2009, 15, 75–83. [Google Scholar] [CrossRef]
- Ruppert, C.; Deiss, K.; Herrmann, S.; Vidal, M.; Oezkur, M.; Gorski, A.; Weidemann, F.; Lohse, M.J.; Lorenz, K. Interference with ERK(Thr188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 7440–7445. [Google Scholar] [CrossRef]
- Vidal, M.; Wieland, T.; Lohse, M.J.; Lorenz, K. β-Adrenergic receptor stimulation causes cardiac hypertrophy via a Gβγ/Erk-dependent pathway. Cardiovasc. Res. 2012, 96, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Brietz, A.; Schuch, K.V.; Wangorsch, G.; Lorenz, K.; Dandekar, T. Analyzing ERK 1/2 signalling and targets. Mol. Biosyst. 2016, 12, 2436–2446. [Google Scholar] [CrossRef]
- Lee, J.D.; Ulevitch, R.J.; Han, J.H. Primary structure of BMK1: a new mammalian MAP Kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [Google Scholar] [CrossRef]
- Kimura, T.E.; Jin, J.; Zi, M.; Prehar, S.; Liu, W.; Oceandy, D.; Abe, J.; Neyses, L.; Weston, A.H.; Cartwright, E.J.; et al. Targeted deletion of the extracellular signal-regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload-induced apoptosis in the heart. Circ. Res. 2010, 106, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.M.; Kuo, W.W.; Yang, J.J.; Wang, S.G.P.; Yeh, Y.L.; Tsai, F.J.; Ho, Y.J.; Chang, M.H.; Huang, C.Y.; Lee, S.D. Eccentric cardiac hypertrophy was induced by long-term intermittent hypoxia in rats. Exp. Physiol. 2007, 92, 409–416. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, W.; Geng, J.; Wang, L.; Su, G.; Zhang, Y.; Ge, Z.; Kang, W. Protein kinase C epsilon-dependent extracellular signal-regulated kinase 5 phosphorylation and nuclear translocation involved in cardiomyocyte hypertrophy with angiotensin II stimulation. J. Cell. Biochem. 2009, 109, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.J.; Itoh, S.; Baines, C.P.; Zhang, C.; Ohta, S.; Che, W.; Glassman, M.; Lee, J.D.; Yan, C.; Yang, J.; et al. Activation of big MAP kinase 1 (BMK1/ERK5) inhibits cardiac injury after myocardial ischemia and reperfusion. FEBS Lett. 2004, 566, 255–260. [Google Scholar] [CrossRef]
- Chen, X.; Cai, H.; Chen, Q.; Xie, H.; Liu, Y.; Lu, Q.; Tang, Y. Effects of Wenyangzhenshuai Granule on ERK1/2 and ERK5 activity in the myocardial tissue in a rabbit model of adriamycin-induced chronic heart failure. Int. J. Clin. Exp. Med. 2015, 8, 20732–20741. [Google Scholar] [PubMed]
- Itoh, N.; Ohta, H.; Nakayama, Y.; Konishi, M. Roles of FGF signals in heart development, health, and disease. Front. Cell. Dev. Biol. 2016, 4, 110. [Google Scholar] [CrossRef]
- Arechederra, M.; Carmona, R.; González-Nuñez, M.; Gutiérrez-Uzquiza, Á.; Bragado, P.; Cruz-González, I.; Cano, E.; Guerrero, C.; Sánchez, A.; López-Novoa, J.M.; et al. Met signaling in cardiomyocytes is required for normal cardiac function in adult mice. Biochim. Biophys. Acta Mol. Basis. Dis. 2013, 1832, 2204–2215. [Google Scholar] [CrossRef]
- Schreier, B.; Rabe, S.; Schneider, B.; Bretschneider, M.; Rupp, S.; Ruhs, S.; Neumann, J.; Rueckschloss, U.; Sibilia, M.; Gotthardt, M.; et al. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension 2013, 61, 333–340. [Google Scholar] [CrossRef]
- Sysa-Shah, P.; Xu, Y.; Guo, X.; Belmonte, F.; Kang, B.; Bedja, D.; Pin, S.; Tsuchiya, N.; Gabrielson, K. Cardiac-specific over-expression of epidermal growth factor receptor 2 (ErbB2) induces pro-survival pathways and hypertrophic cardiomyopathy in mice. PLoS ONE 2012, 7, e42805. [Google Scholar] [CrossRef]
- Cilvik, S.N.; Wang, J.I.; Lavine, K.J.; Uchida, K.; Castro, A.; Gierasch, C.M.; Weinheimer, C.J.; House, S.L.; Kovacs, A.; Nichols, C.G.; et al. Fibroblast growth factor receptor 1 signaling in adult cardiomyocytes increases contractility and results in a hypertrophic cardiomyopathy. PLoS ONE 2013, 8, e82979. [Google Scholar] [CrossRef]
- Sala, V.; Gallo, S.; Gatti, S.; Medico, E.; Vigna, E.; Cantarella, D.; Fontani, L.; Natale, M.; Cimino, J.; Morello, M.; et al. Cardiac concentric hypertrophy promoted by activated Met receptor is mitigated in vivo by inhibition of Erk1,2 signalling with Pimasertib. J. Mol. Cell. Cardiol. 2016, 93, 84–97. [Google Scholar] [CrossRef]
- Leo, C.; Sala, V.; Morello, M.; Chiribiri, A.; Riess, I.; Mancardi, D.; Schiaffino, S.; Ponzetto, C.; Crepaldi, T. Activated met signling in the developing mouse heart leads to cardiac disease. PLoS ONE 2011, 6, e14675. [Google Scholar] [CrossRef] [PubMed]
- Kagiyama, S.; Eguchi, S.; Frank, G.D.; Inagami, T.; Zhang, Y.C.; Phillips, M.I. Angiotensin II-induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation 2002, 106, 909–912. [Google Scholar] [CrossRef]
- Peng, K.; Tian, X.; Qian, Y.; Skibba, M.; Zou, C.; Liu, Z.; Wang, J.; Xu, Z.; Li, X.; Liang, G. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J. Cell. Mol. Med. 2016, 20, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, X.; Kong, M.; Jiang, D.; Dong, A.; Shen, Z.; Duan, Q. Cardiac-targeting magnetic lipoplex delivery of SH-IGF1R plasmid attenuate norepinephrine-induced cardiac hypertrophy in murine heart. Biosci. Rep. 2014, 34, e00140. [Google Scholar] [CrossRef]
- Pires, K.M.; Buffolo, M.; Schaaf, C.; David Symons, J.; Cox, J.; Abel, E.D.; Selzman, C.H.; Boudina, S. Activation of IGF-1 receptors and Akt signaling by systemic hyperinsulinemia contributes to cardiac hypertrophy but does not regulate cardiac autophagy in obese diabetic mice. J. Mol. Cell. Cardiol. 2017, 113, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Alcalai, R.; Seidman, J.G.; Seidman, C.E. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J. Cardiovasc. Electrophysiol. 2008, 19, 104–110. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef]
- Patel, R.; Nagueh, S.F.; Tsybouleva, N.; Abdellatif, M.; Lutucuta, S.; Kopelen, H.A.; Quinones, M.A.; Zoghbi, W.A.; Entman, M.L.; Roberts, R.; et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001, 104, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Liu, X.; Sparrow, A.; Patel, S.; Zhang, Y.H.; Casadei, B.; Watkins, H.; Redwood, C. Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J. Biol. Chem. 2018, 293, 10487–10499. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Davis, L.C.; Correll, R.N.; Makarewich, C.A.; Schwanekamp, J.A.; Moussavi-Harami, F.; Wang, D.; York, A.J.; Wu, H.; Houser, S.R.; et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [Google Scholar] [CrossRef]
- Higgins, E.M.; Bos, J.M.; Mason-Suares, H.; Tester, D.J.; Ackerman, J.P.; MacRae, C.A.; Sol-Church, K.; Gripp, K.W.; Urrutia, R.; Ackerman, M.J. Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight 2017, 2, e91225. [Google Scholar] [CrossRef] [PubMed]
- Kontaridis, M.I.; Yang, W.; Bence, K.K.; Cullen, D.; Wang, B.; Bodyak, N.; Ke, Q.; Hinek, A.; Kang, P.M.; Liao, R.; et al. Deletion of Ptpn11 (Shp2) in cardiomyocytes causes dilated cardiomyopathy via effects on the extracellular signal-regulated kinase/mitogen-activated protein kinase and RhoA signaling pathways. Circulation 2008, 117, 1423–1435. [Google Scholar] [CrossRef]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef]
- Kobayashi, T.; Aoki, Y.; Niihori, T.; Cavé, H.; Verloes, A.; Okamoto, N.; Kawame, H.; Fujiwara, I.; Takada, F.; Ohata, T.; et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum. Mutat. 2010, 31, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Simpson, J.; Hong, J.H.; Kim, K.H.; Thavarajah, N.K.; Backx, P.H.; Neel, B.G.; Araki, T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 2011, 121, 1009–1025. [Google Scholar] [CrossRef]
- Li, C.; Chen, Z.; Yang, H.; Luo, F.; Chen, L.; Cai, H.; Li, Y.; You, G.; Long, D.; Li, S.; et al. Selumetinib, an oral anti-neoplastic drug, may attenuate cardiac hypertrophy via targeting the ERK pathway. PLoS ONE 2016, 11, e0159079. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Extrinsic Stimuli | |||
|---|---|---|---|
| Experimental Models | Response | References | |
| Pressure Overload | Transverse aortic constriction (TAC) in mice | Early adaptive concentric hypertrophy; ERK ↑; Late detrimental eccentric hypertrophy; ERK ↓ | [19] |
| Aortic Valve Stenosis | Human patients | Detrimental eccentric hypertrophy; ERK ↓ | [87] |
| AngII | Cardiomyocytes | ERK5 ↑; ERK ↑ | [10,93,104] |
| AngII inhibitors | Heart failure patients | Reduction of cardiac hypertrophy and heart failure; ERK ↓ | [24] |
| Isopreteronol | βAR stimulation in mice | Cardiac hypertrophy and fibrosis; ERK phosphorylation at T188 ↑ | [88] |
| βAR Blockers | In vitro treatment | Arrestin-mediated EGF receptor transactivation; ERK ↑ | [28,29] |
| Anthracycline | In vitro and in vivo treatments | Heart failure; ERK ↑ | [71,72] |
| In vitro and in vivo treatments | Cardioprotective action; ERK ↑ | [74,75,76] | |
| Rat cardiomyocytes (Lapatinib and Doxorubicin) | Cardiotoxicity; ERK ↓ | [84] | |
| Trastuzumab | Human cardiomyocytes | Cardiotoxicity; ERK ↑ | [85] |
| Intrinsic Stimuli | |||
| Experimental Models | Response | References | |
| DN RAF-1 | Cardiomyocytes-specific Tg mice | Blunted response to pathological hypertrophy; ERK ↓ | [30] |
| DUSP-6 | Cardiomyocytes-specific Tg mice | Heart failure in response to TAC; ERK ↓ | [32] |
| MEK1 | Cardiomyocytes-specific Tg mice | Concentric cardiac hypertrophy; ERK ↑ | [4] |
| MEK5β | Cardiomyocytes-specific Tg mice | Eccentric cardiac hypertrophy and heart failure; ERK5 ↑ | [10] |
| MEK5α | Cardiomyocytes-specific Tg mice | Prevention of heart failure in response to MI; ERK5 ↑ | [94] |
| ERK5 | Cardiomyocytes-specific knock out mice | Reduced cardiac hypertrophy, and increased apoptosis in response to TAC; ERK5 ↓ | [91] |
| αAR | α(1A/C)AR and α(1B)AR double knock out mice | Small heart with reduced cardiac output in response to TAC; ERK ↓ | [34] |
| α1AR | knock out mice | Pathological hypertrophy and heart failure in response to MI; ERK ↓ | [35] |
| β1AR | Cardiomyocytes-specific Tg mutant mice | Lack of EGFR transactivation; Increased contractility, fibrosis and apoptosis; ERK ↓ | [28] |
| βArrestin | In vitro knock out | Arrestin 1: ERK ↑; Arrestin 2: ERK ↓ | [21] |
| βArrestin | Knock out mice | Lack of EGFR transactivation; ERK ↓ | [28,29] |
| Erbin | Knock out mice | Cardiac hypertrophy and heart failure in response to TAC; ERK ↓ | [42] |
| IQGAP1 | Knock out mice | Eccentric hypertrophy in response to TAC; ERK ↓ | [44] |
| Melusin | Cardiomyocytes-specific Tg mice | Concentric hypertrophy, improved response to TAC; ERK ↑ | [47] |
| FHL1 | Knock out mice | Blunted response to pathological hypertrophy; ERK ↓ | [48] |
| ANKRD1 | Cardiomyocytes knock down and knock out mice | Blunted response to pathological hypertrophy; ERK ↓ | [51] |
| ERK2 T188A | Cardiomyocytes-specific Tg mice | Attenuation of pathological hypertrophy in response to GPCRs activation and TAC | [87,88] |
| HGFR | Cardiomyocytes-specific Tg mice | Early adaptive concentric hypertrophy; late heart failure; ERK ↑ | [101] |
| EGFR | In vitro and in vivo protein knock down | Failure of AngII-mediated cardiac hypertrophy; ERK ↓ | [103,104] |
| IGF1R | Cardiomyocytes-specific protein knock down in mice | Attenuation of norepinephrine-induced cardiac hypertrophy; ERK ↓ | [105] |
| HCM | βMHC-Q(403) in Tg rabbits | Cardiac hypertrophy, fibrosis, and contractile dysfunction; ERK ↑ | [110] |
| cTnT R92Q, cTnI R145G, and αTM D175N in cardiomyocytes | Cardiomyocyte hypertrophy; ERK ↑ | [111] | |
| I61Q cTnC in cardiomyocytes | Failure of ERK translocation to the nucleus and cardiomyocytes elongation | [112] | |
| R193H cTnI in cardiomyocytes | ERK translocation to the nucleus and increased cardiomyocytes width | [112] | |
| RASopathies | Cardiomyocytes-specific knock out of PTPN11 in mice | Failure in the induction of adaptive hypertrophy; ERK ↓ | [114] |
| Noonan RAF-1 L613V mutation knock in mice | Eccentric hypertrophy and heart failure; ERK ↑ | [117] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 2164. https://doi.org/10.3390/ijms20092164
Gallo S, Vitacolonna A, Bonzano A, Comoglio P, Crepaldi T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. International Journal of Molecular Sciences. 2019; 20(9):2164. https://doi.org/10.3390/ijms20092164
Chicago/Turabian StyleGallo, Simona, Annapia Vitacolonna, Alessandro Bonzano, Paolo Comoglio, and Tiziana Crepaldi. 2019. "ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy" International Journal of Molecular Sciences 20, no. 9: 2164. https://doi.org/10.3390/ijms20092164
APA StyleGallo, S., Vitacolonna, A., Bonzano, A., Comoglio, P., & Crepaldi, T. (2019). ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. International Journal of Molecular Sciences, 20(9), 2164. https://doi.org/10.3390/ijms20092164