In the following flow chart, we summarize the analysis flow and how the results build on each other.
2.1. ERK is of Pivotal Significance in the Cardiomyocyte’s Regulatory Network
ERK1/2 have shown to be essential but at the same time detrimental to the heart: ERK1/2 mediate cell survival but can also mediate cardiomyocyte hypertrophy associated with maladaptive remodeling of the heart and impaired cardiac function. The selective prevention of ERK1/2-mediated cardiac hypertrophy—but not of ERK1/2-mediated cell survival—is thus of interest for the prevention of heart failure [
6].
In this section, we consider a gene regulatory network for cardiomyocytes given in [
14] where we discuss how to use the presented framework in principle to calculate different strategies to act on this network with external stimuli and to find out optimal pharmacological targets. A strategy is defined as which nodes we have to activate or inhibit by external stimuli. The network’s graph is shown in
Figure 2. Here, we would like to make another point: A model is never accurate and hence there are always additions you can consider in a more complex model. For example, Epac does not seem to activate PKC, but rather it can activate Ras. However, we start with a simpler model as we aim to give a general framework for network and drug target analyzing. On the one hand, the framework is for any model that is set up of differential equations and not fixed to the model depicted in
Figure 2. For a practical use, we provide the corresponding Matlab scripts where the user just has to change the corresponding model equations that can also be generated with Jimena [
15], SQUAD [
16] or Potterswheel [
17].
Hence, for explaining the ERK cascade, we use the underlying mathematical model (2.1) in the
Supplementary Materials if not otherwise stated where
and
,
, which are taken from [
14] and are given as in
Table 1. The nodes AND and SYSTEM STATE are technical nodes, which we describe in the following. The node SYSTEM STATE is not in the network but is used to permanently activate RKIP and GRK2 to model their constitutive expression in our model of a cardiomyocyte. Furthermore, the node SYSTEM STATE activates the node AND such that we have that ERK1/2 dim 3P can only be activated if ERK1/2 dim and
is active at the same time since the three times phosphorylated ERK1/2 requires the twice phosphorylated ERK1/2. This models the fact that the interdependence between ERK1/2 dimer with two phosphorylations from the positive inotropic cascade and the ERK1/2 dimer with three phosphorylations is represented by an “AND” connection. The equations for node 1, node 10 and node 26 are given as
,
, which can be supplemented by activating stimuli according to model (2.1) in the
Supplementary Materials and
that ensures that the activity level of SYSTEM STATE is constantly one, which means is set on, to activate node 14, node 16 and node 17.
The control strategy is defined for each experiment separately. We have for the parameters if the external stimulus has no effect on the node k, if the external stimulus has an activating effect on the node k and if external stimulus has an inhibiting effect on node k. It is stated in this work if we use different values for the parameters than these ones. Thus, now we should consider pharmacological knowledge to think about optimal therapy strategies regarding the ERK cascade:
In our case, we associate a high activity of the nodes p90RSK (node 8) and p70S6K (node 9) with beneficial effects and a high activity of the nodes Elk1 (node 23), MSK1 (node 24) and c-Myc (node 25) with maleficent effects.The activity levels of nodes, which ranges in our work between zero and one, stand for the biological production activities of the associated biological agents. For instance, transcription or translation rates of the associated node is gene. In this case, zero is associated with no production and is interpolated until one which represents the highest production rate that is biologically possible.
From our considerations, we desire a high activity for p90RSK and p70S6K and a low activity for Elk1, MSK1 and c-Myc. We define these five nodes as our nodes of interest and choose the desired state for the first two ones constant one and for the last three constant zero. The weights
for the first two are equal to
for the other three equal to 1 to compensate the fact that we have two beneficial nodes and three maleficent ones and thus give the beneficial effect altogether the same weight as the maleficent effect. For our experiments, we always had
the constant zero vector except the last entry was set to 1, which is the initial value of the system state. To calculate the pharmacological intervention in order to obtain the desired activity pattern of the nodes, we used Algorithm 1 in the
Supplementary Materials where we set its parameters as follows
. If we used fewer than 10 possible external stimuli, which are the intervention possibilities in the lab, then we set
to at most the number of used external stimuli. Furthermore, we set
. The result from these calculations can be used as an initial guess for the sequential Hamiltonian (SQH) method [
18] (Algorithm 2) to calculate the fine-tuning for the external stimuli. For this purpose, we use the recommended parameter values from [
18] except
and
if not otherwise stated. The final time is chosen by
which means that the simulation time for the regulatory network is 20 time units. We compare pharmacological treatment strategies as follows.
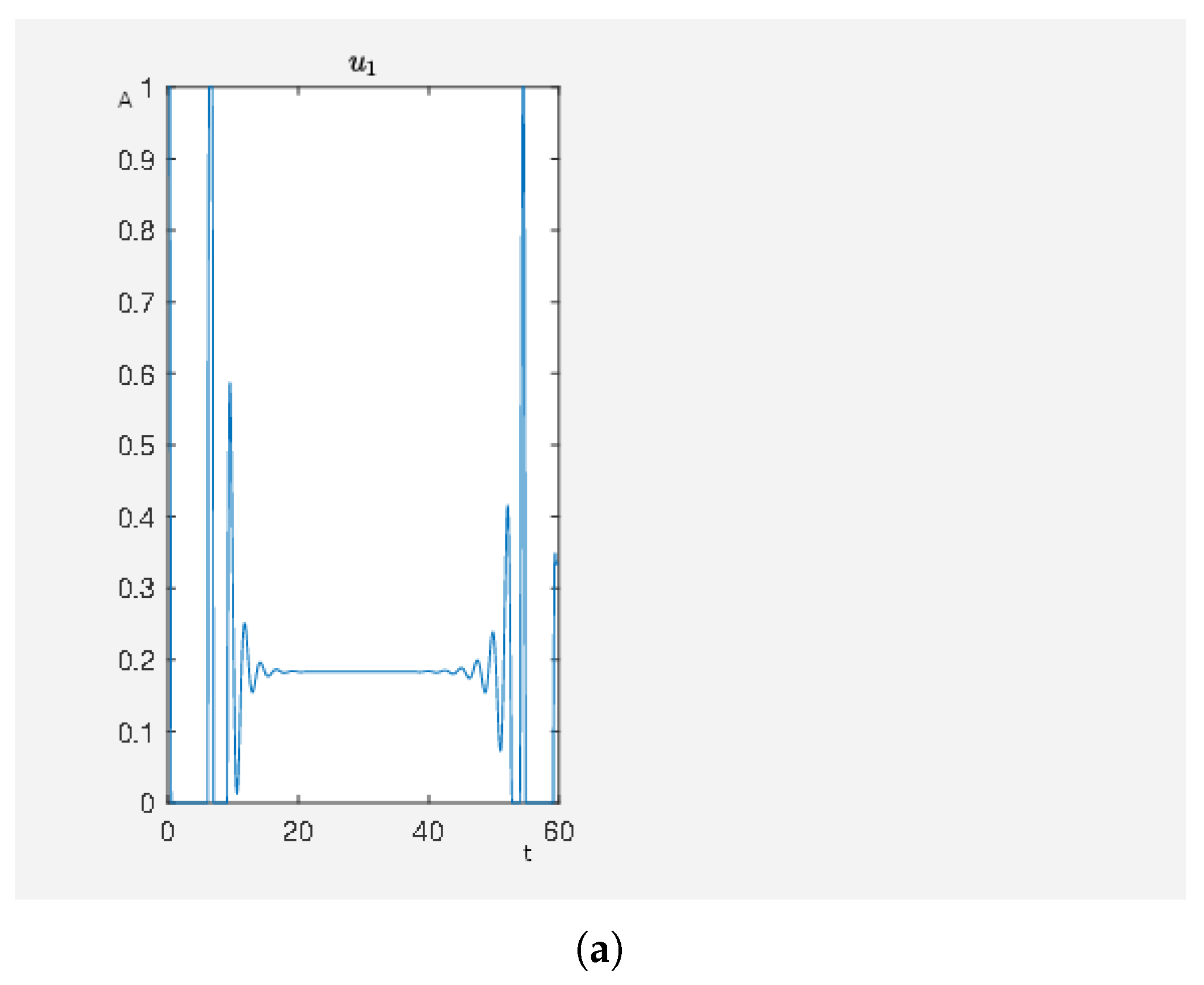
For our first experiment, we wanted to study the effects of carbachol, angiotensin II and isoproterenol. These are drugs typically used in the clinic to treat high blood pressure (angiotensin 1 receptor blockers) and heart failure (beta receptor blockade) and are, in particular in older, multi-morbid patients, often used in combination. Thus, translated into the mathematical language of our framework, this reads as follows.
We have an activating external stimulus on the node carbachol, angiotensin II and isoproterenol. When the SQH method stops and returns a solution, we have
and, in
Figure 3, we can see the time curves of the external stimuli, which are not the zero function and the time curves of the states of interest. We see that an activation of carbachol leads to the activation of the beneficial nodes. The short pulse of angiotensin II supports this effect and the maleficent nodes decay after a short and weak activation.
On the other hand, our framework can be used to check if a model is reasonable by testing what happens subject to different external stimuli and if the reactions are in correspondence to experimental data. Remember, the phosphorylation of p70S6K by ERK does not cause the direct activation of the latter and further regulatory events may activate p70S6K. However, everything can be modeled in more detail, considering further or alternative regulatory events. In fact, which important effects to consider depends on the choice of the user. The effects in
Figure 3 are a result of the calculation to steer the network depicted in
Figure 2 to the desired state. We aimed as follows. By our straightforward framework, we can evaluate models by checking if external stimuli provide known results. In this way, we can easily check if a model is reasonable by testing different situations of external stimuli and their corresponding effects. A more detailed model would refine the model output shown in
Figure 3.
2.1.1. Studying the Effects of Mono-Therapy on the ERK Signaling Network
In
Figure 4, we see the result where we only have an activating external stimulus on carbachol. The target functional value
when the SQH method converges. If we compare the target functional value with the first experiment, we see that it is just a bit bigger and thus both control strategies can be seen as equivalent control strategies with respect to an activity level close to the desired one.
In our third experiment, we had an activating external stimulus just on angiotensin II and isoproterenol. This would correspond to a simultaneous pharmacological treatment with angiotensin II and isoproterenol. When the SQH method converges, we have
. In
Figure 5, we have the time curves of the external stimuli which are not zero and of the nodes of interest. Compared with the two other experiments, the target functional was much higher, which means that an activating external stimulus on carbachol was essential for an activity level of the network’s nodes of interest close to our desired activity level. By this, we would then avoid heart hypertrophy and generate a strong non-hypertrophic stimulus though.
2.1.2. Combined Effects of Activation and Inhibition on ERK Signaling
ERK autophosphorylation triggers cardiac hypertrophy and subsequent maladaptive remodeling and cardiac insufficiency. Since modulation of ERK phosphorylation does not affect ERK1/2 mediated cell survival, ERK phosphorylation is thought to be an elegant target to intervene with maladaptive ERK1/2 signaling in the heart.
In our fourth experiment, we had activating external stimuli on angiotensin II and isoproterenol and one inhibiting external stimulus on ERK1/2 dim 3P. When the SQH method converges, we have
and the corresponding time curves are shown in
Figure 6. As the target functional value is close to the one with the experiments where carbachol is activated, we can say that the strategy of activating angiotensin II and isoproterenol while inhibiting ERK1/2 dim 3P is equivalent to the one where we only activate carbachol.
In
Figure 7a, we have the activity level of ERK1/2 dim 3P, which is of course an extreme case as we have a very strong inhibition. However, it demonstrates that for a strong inhibition of the ERK1/2 dim 3P that this strategy is almost that good as using an activating stimulus only on carbachol, see the first two experiments.
It is possible to fit an external stimulus’s ability for inhibition by the parameter
in (2.1) of the
Supplementary Materials such that the corresponding node has the measured activity level when the inhibitor is active. For this purpose, the parameter
can be diminished and thus the activity level of ERK1/2 dim 3P increases for
tending to zero, see
Figure 7b.
These first four experiments demonstrated how our optimization framework can be used to compare different control strategies with respect to their ability to steer the activity level to the desired activation level of the network’s nodes. Once the parameters such as
T and weights
are fixed (see model (2.2) in the
Supplementary Materials), then the smaller the target functional value of a certain control strategy is, the more beneficial effects and the less maleficent effects the strategy has. By this procedure, we can sort different strategy or assess them with respect to their corresponding target functional value. The optimization framework serves as an objective method to determine the time curves of the external stimuli such that we have the lowest target functional value possible for the given strategy. We stress that we just influence on which node an external stimuli acts. Each time curve is then automatically given by the optimization framework, namely by solving model (2.2) in the
Supplementary Materials.
In the next two sections, we show how to use our framework to model constitutively activated signal pathways since they play an important role in heart failure and for oncogenesis in general. Then, in the following, we show how to utilize the framework to systematically search for optimal treatment strategies, again with our basal model of a cardiomyocyte.
2.2. A Cardiomyocyte with Constitutively Activated -Coupled Receptor
This situation can arise from different situations in real life: For instance, taking constantly a beta-mimetic drug to treat asthma could lead to such a constant activation of the
-coupled
receptor. Alternatively, endogenous factors such as constant stress or first signs of cardiac failure can lead to such an activation. Mathematically speaking, we hence discuss different strategies for the network from
Section 2.1 where the
-coupled
receptor (node 19) is constitutively activated such that it has continuously about
of its maximum activation level. To model this, we equip its corresponding activating node isoproterenol (node 18) with the term
such that the corresponding equation is given by
. Furthermore, in our experiment, node 10 (hypertrophic stimulus) was not activated and thus stayed at zero if an initial value of zero was chosen. This ensured that isoproterenol stayed at a constant level of
of its maximum activation, which had the consequence that the
-coupled
receptor had about
of its maximum activation level (see
Figure 8). As in
Section 2.1, we associate a high activity of the nodes p90RSK (node 8) and p70S6K (node 9) with beneficial effects and a high activity of the nodes Elk1 (node 23), MSK1 (node 24) and c-Myc (node 25) with maleficent effects, which is why we desire a low activity for them. We define these five nodes as our nodes of interest and choose the desired state for the first two constants one and for the last three constants zero. The weights
for the the first two are again equal to
for the other three equal to 1. We always have
equal to the constant zero vector except the activity level for AND, GRK2 and SYSTEM STATE equal 1 and we use Algorithm 1 in the
Supplementary Materials where we set the algorithm’s parameters as follows
and
. The result from this calculation was used as the initial guess for the sequential Hamiltonian (SQH) method [
18] (Algorithm 2) to calculate the fine tuning for the relevant intervention points. The SQH method was used with its recommended parameter values, see [
18], except
and
if not otherwise stated. The final time was chosen by
, i.e., the regulatory network was simulated for 60 time units.
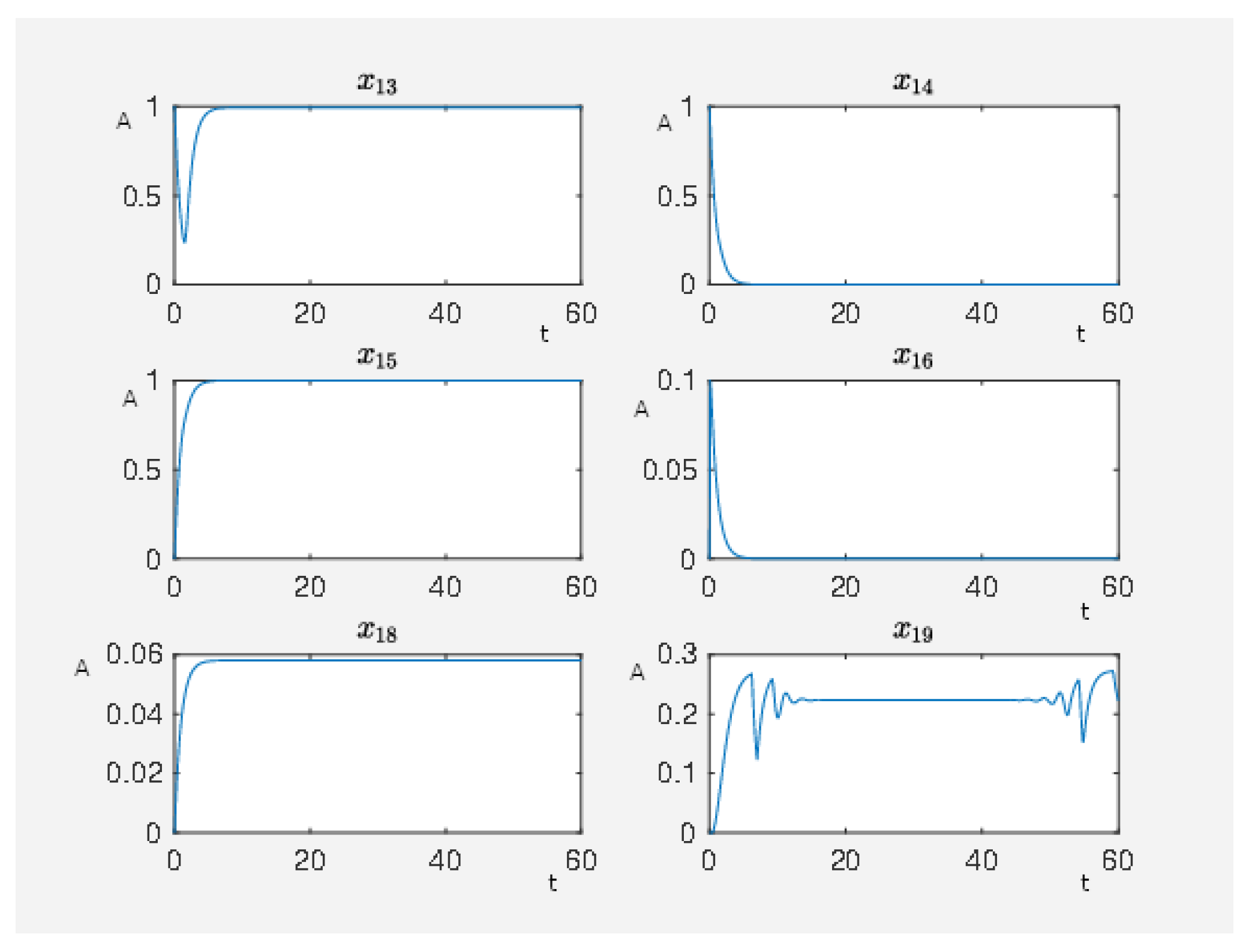
If the network is unperturbed, then the constitutively activated
-coupled
receptor causes the following activity pattern in the network, where we show the activity level of some nodes in
Figure 8 with
. We see that the activity level of the nodes associated with maleficent effects (nodes 23–25) are highly active while the nodes associated with beneficial effects (nodes 8 and 9) are at a very low activity level. Furthermore, we see that a constitutively activated receptor is able to hold the network in a certain state that means a constant expression pattern. Thus, the expression pattern of the network is also constitutively altered compared to the steady state in which the network would be if the receptor was totally inactive.
2.2.1. Further Pathological Constant Molecular Activation
In this subsection, we would like to look at the long-term consequences of the constant activation of the ERK cascade by the constitutively activated -coupled receptor. In particular, the continuous activation of a receptor or ERK kinase or other members of the ERK cascade such as MEK may also lead to cancer.
This can also easily be investigated within our framework. In such situations, ERK is part of the signaling cascade while the constitutive activation may either stem from an activating, oncogenic mutation of a key receptor such as Epidermal Growth Factor Receptor (EGFR, usually treated by Gefitinib [
19]) or by a kinase mutation (most well known are B-Raf and Ras mutations, however, in some aggressive cancers this can also be ERK mutations).
In general, a constitutively activation of receptors can be caused by mutations in the receptor itself or its corresponding signal protein. Another example is cell–cell-interaction where a constitutively activated receptor can be caused by secretory cells that constitutively secret the corresponding signal molecule. The presented framework in combination with constitutively activated receptors can also be used in modeling oncogenesis where constitutively activated pathways play a role [
20,
21,
22].
A further example is the inhibition of p53 or Retinoblastoma (Rb) protein after a virus infection. This can be caused by constitutively expressed proteins that bind to p53 or Rb to enhance cell proliferation which is needed for the virus reproduction [
23,
24,
25]. This can be modeled analogous to the constitutively activated receptors where the other way round the activity level of the corresponding node is constitutively inhibited by the external stimuli associated with this effect of the virus infection. This illustrates that an external stimulus can also be a virus or the effect of its infection, as well as how this can be modeled within the presented framework. Once the corresponding issue is modeled as described before, the benefit for pharmacological research is the identification of promising drug targets that counter the maleficent effects caused by the constitutively activated pathway. This illustrates how the mechanism modeling constitutively activated
-coupled
receptor as discussed above transfers to different situations and signaling pathways.
2.2.2. Steering the ERK Network of the Cardiomyocyte in a Beneficial Way
Molecular pre-requisites for ERK phosphorylation are the dimerization of ERK and G binding to the ERK dimer. Interference with these protein-protein interactions would thus facilitate selective inhibition of ERK phosphorylation. In this manuscript, we particularly evaluate the outcome of the interference with the ERK–ERK interaction in the context of the different ERK1/2 activating cascades.
Now, we show how to apply our framework to discuss different strategies that improve the expression pattern that means to obtain a non-hypertrophic stimulus. First, we show the effects of just blocking the constitutively activated -coupled receptor and then we demonstrate how to use our framework to automatically search for an alternative treatment strategy.
A strategy to reduce the target functional value, which means that it increases the beneficial effects, is to inhibit the
-coupled
receptor, which is called the
-block strategy in this work. When the SQH method converges, we have
. The results are shown in
Figure 9 and
Figure 10 for some activity levels of nodes. The time curve of the corresponding external stimuli might be a delicate issue in a real experiment. With a constant external stimulus with value
, we have the value of the cost functional
. Therefore, it is not needed to have such a highly structured time curve, as shown in
Figure 9, because we obtain the same order of magnitude of the target function with the corresponding constant external stimulus. Notice that it is sufficient to reduce the activity level of the
-coupled
receptor from about
to about
such that the activity level of Elk1, MSK1 and c-Myc drops from about
to
. That means that we have identified a threshold for the activity of these three nodes via the activation of the G
-coupled
receptor. In this way, we can use the framework to calculate a fine tuning of nodes. By equipping a node by an external stimulus, we see after the calculation how much the activity level upon the action of the external stimulus differs from the unperturbed situation.
2.2.3. Systematic Search for an Optimal Treatment
For clinical applications, steering the ERK signaling pathway is critical. This is achieved usually by pharmacological drugs. However, ERK is in a network and rarely the complete network response is considered. For this reason, we discuss in the following the network effects, too.
To obtain a further improvement of the therapy, i.e., more beneficial effects and fewer side effects, we now perform a systematic search for beneficial intervention points. In mathematical terms, it means that we find external stimuli that effect the activity levels of certain nodes and are associated with a therapy that reduces the target functional, defined in (2.2) in the
Supplementary Materials, which takes maleficent side effects and beneficial effects of a therapy via the corresponding external stimuli into account. In the case of a small target functional value, we have external stimuli that steer the considered network to a state with corresponding activity levels of the nodes that are associated with a healthy state. For our systematic search, we equip the nodes PKC (
), RKIP (
), RKIP dim (
), ERK1/2 dim 3P (
), G
-coupled
receptor (
) and Ras (GTP bound) (
) with inhibiting external stimuli and the nodes angiotensin II (
) and isoproterenol (
) with activating external stimuli. In our example, we set
to model the fact that possibly the external stimuli
cannot totally inactivate of ERK1/2 dim 3P. In
Figure 11, we can see the results. Notice that we now have high activity levels for the nodes p90RSK and p70S6K and still a low activity level for the nodes Elk1, MSK1 and c-Myc which results in a lower target functional value than in the last experiment. We have a target functional value
. That means this treatment is better than just blocking the G
-coupled
receptor. However, there are many external stimuli active. Our next step is to use our framework to reduce the number of external stimuli in order to obtain the most effective external stimuli. These are the external ones to focus on for designing an optimal therapy.
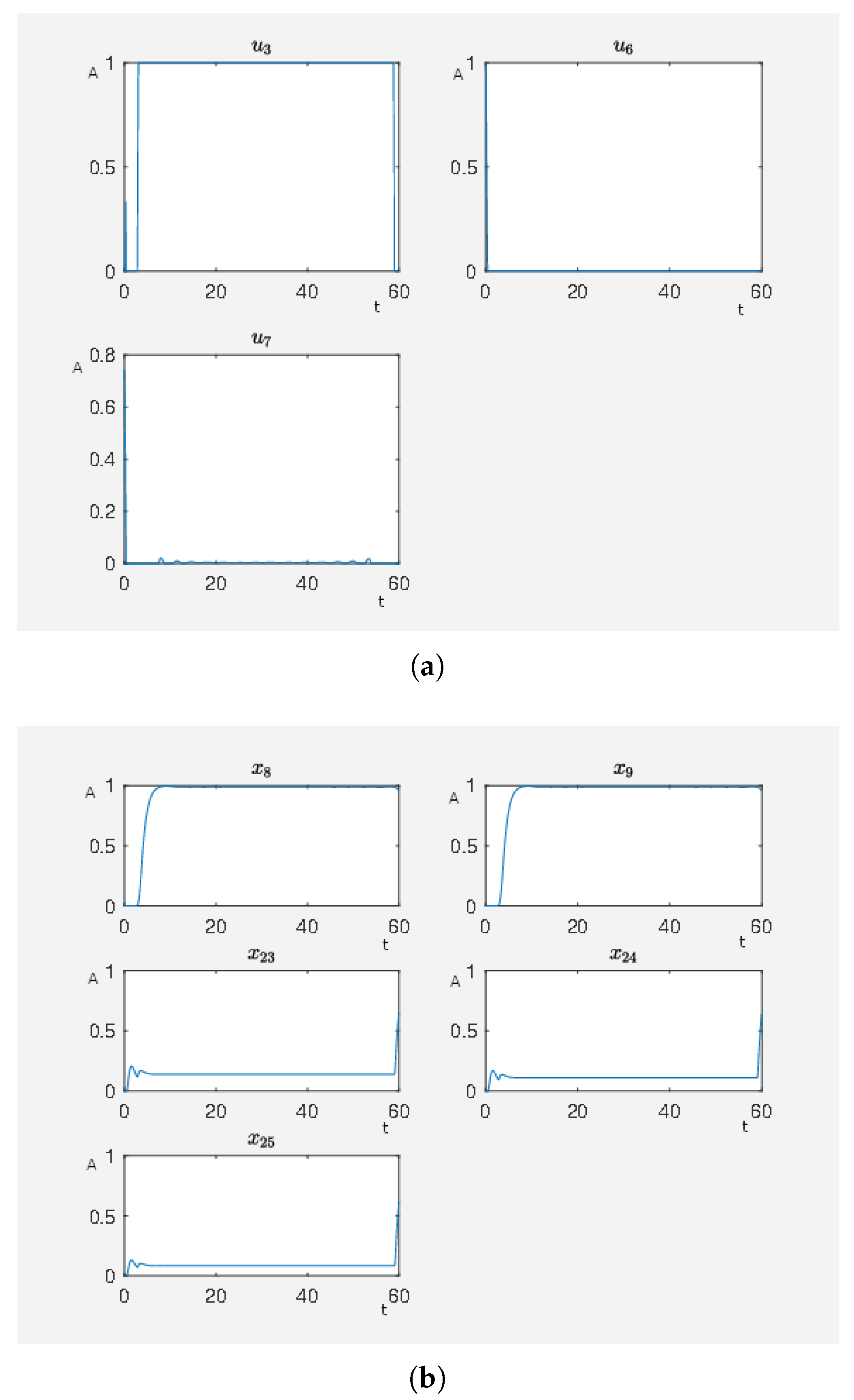
In
Figure 11, there are many active external stimuli. By increasing
in model (2.2) of the
Supplementary Materials, we reduce the number of active external stimuli. This comes from the fact that by increasing alpha the costs for active external stimuli increase such that only that ones remain whose activity has a noteworthy effect on steering to network to our desired expression pattern. Thus, we extract the most effective external stimuli which have much effect on reducing the target functional value and our framework returns only these external stimuli that are really important. We increase
and perform the calculations where, for
, we only have
,
and
as active external stimuli. The results can be seen in
Figure 12. This means that these three external stimuli, which are
inhibits ERK1/2 dim 3P,
activates angiotensin II and
activates isoproterenol, are the important ones that we further investigate.
If we perform the same experiment just with the the active stimuli from
Figure 12 for
where we only use our combinatorial method (see Algorithm 1 in our
Supplementary Materials), we then obtain
for a full activity of
and
(activity level of G
-coupled
receptor about
of its maximum activation level) and
for full activity of
and
(activity level of G
-coupled
receptor about
of its maximum activation level) which is almost the same target functional value as in the case with many external stimuli more shown in
Figure 11. This demonstrates that this combination of external stimuli
and
or
and
are the essential ones to obtain a beneficial effect on the network, which means to have a low target functional value. While the other external stimuli also have beneficial effects, their contributions are minor compared to the effects of
and
or
and
. We can say that compared with the value of the target functional of the unperturbed system (
the strategies
and
or
and
are equivalent with the strategy shown in
Figure 11 where the big advantage is that only two stimuli have to be applied instead of seven.
We remark without the external stimulus , we are just able to obtain a target functional value of , which stresses the importance of the inhibition of ERK1/2 dim 3P for achieving of a beneficial state for the network.
2.2.4. A Threshold for ERK Signaling
Next, we look at the sensitivity of the ERK signaling pathway. We investigate to what activity level ERK1/2 dim 3P has to be knocked down, i.e., has to be reduced, in order to be still as good as just inhibiting
-coupled
receptor, that means to obtain a target functional value
. We take the control strategy that
inhibits ERK1/2 dim 3P and
activates angiotensin. For this purpose, we use our combinatorial method (see Algorithm 1 in our
Supplementary Materials), for different values of
which models the strength how much the activity level of ERK1/2 dim 3P (node 22) can be inhibited by the external stimulus
. The results are presented in
Table 2, where for each experiment the external stimuli are fully active. We see that if the activity level of ERK1/2 dim 3P is at least at
of its maximum activity then, we still have a small target functional value
compared with
which is achieved by just applying our
-block strategy mentioned above. Furthermore, as the activity level of p90RSK and p70S6K are at 1 for all
in
Table 2, we have that at about
of the maximum activity level of ERK 1/2 dim 3P the maleficent effects abruptly increase (activity levels of Elk1, MSK1 and c-Myc) which can be associated with an abrupt worsening of the treatment. For example, if one has a further restriction such as that the activity level of Elk1, MSK1 and c-Myc is supposed to be below
, then one can see from
Table 2 that the activity level of ERK 1/2 dim 3P is supposed to stay below
of its maximum activity level. This can also be seen in
Figure 13 where
abruptly increases at
of ERK12 dim 3P maximum activity level. The interpretation is that the treatment abruptly worsens at
of ERK1/2 dim 3P maximum activity level. However, it does not mean that the treatment is already worse since higher values of
can be tolerable in vivo such that only the beneficial effects are present while the maleficent effects are still not noticeable.
Additionally we still have a high activity level of p90RSK and p70S6K in contrast the strategy where one inhibits just the G
-coupled
receptor. Furthermore, we see in
Table 2 that the more we are able to inhibit ERK1/2 dim 3P the better it is for the treatment, i.e., the lower are the activity levels of Elk1, MSK1 and c-Myc.
In
Figure 14, we can see the corresponding time curve for the activity level of ERK1/2 dim 3P at
of its maximum level. In this case, the activity level of p90RSK and p70S6K is 1 and of Elk1, MSK1 and c-Myc is between
and
. We conclude that if the activity levels of p90RSK and p70S6K are low in spite of a constitutively activated G
-coupled
receptor, one can recommend to activate the G
-coupled
receptor even more by angiotensin II until the activity levels of p90RSK and p70S6K are high if one can manage it at the same time to inhibit the third phosphorylation of ERK1/2.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}