The Pathogenesis of Port Wine Stain and Sturge Weber Syndrome: Complex Interactions between Genetic Alterations and Aberrant MAPK and PI3K Activation



Abstract
:1. Introduction
2. Clinical Background of PWS/SWS
3. Pathological Phenotypes of PWS/SWS
3.1. Ultrastructure of PWS Lesions
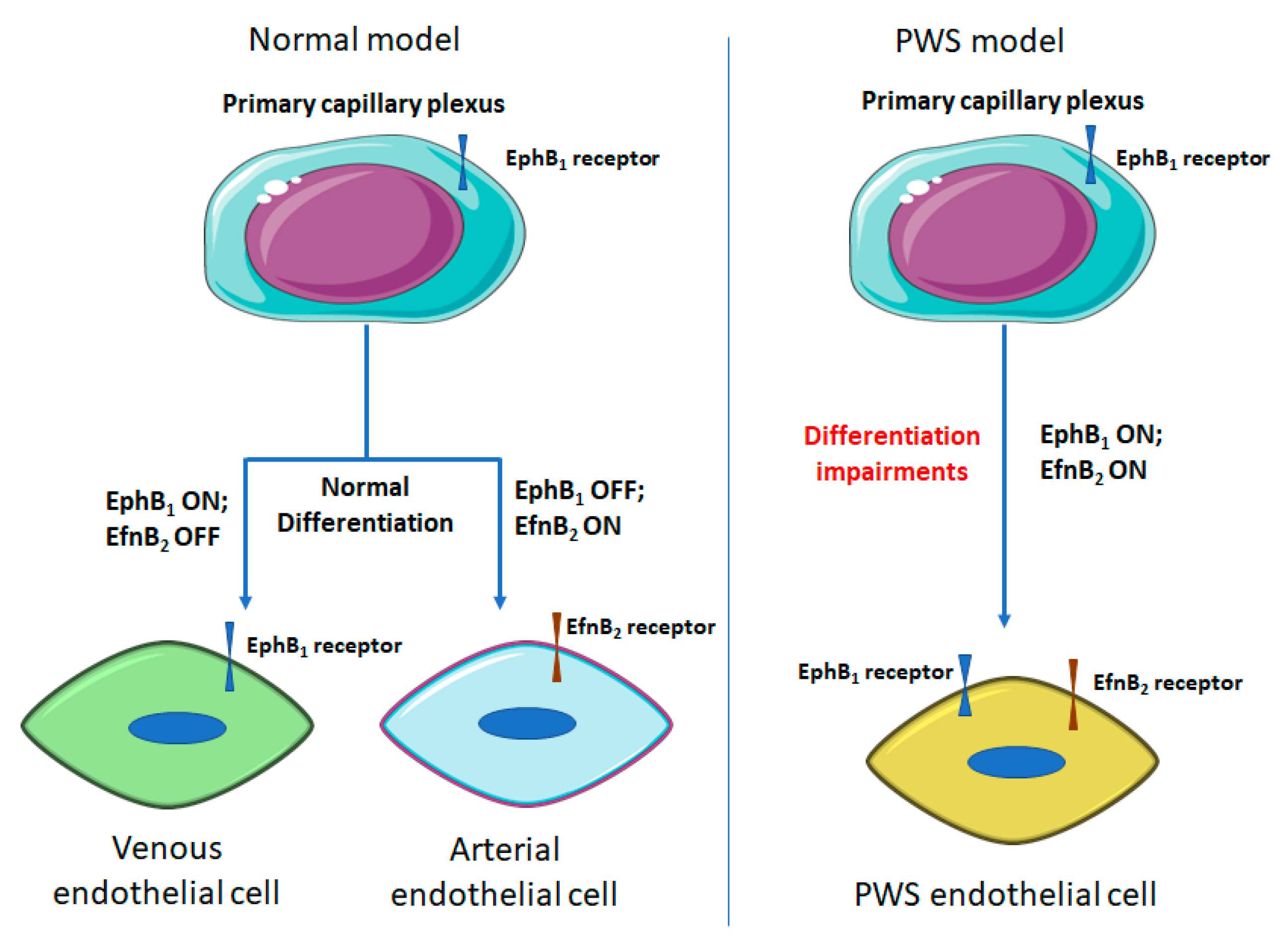
3.2. Differentiation Impairments of PWS ECs
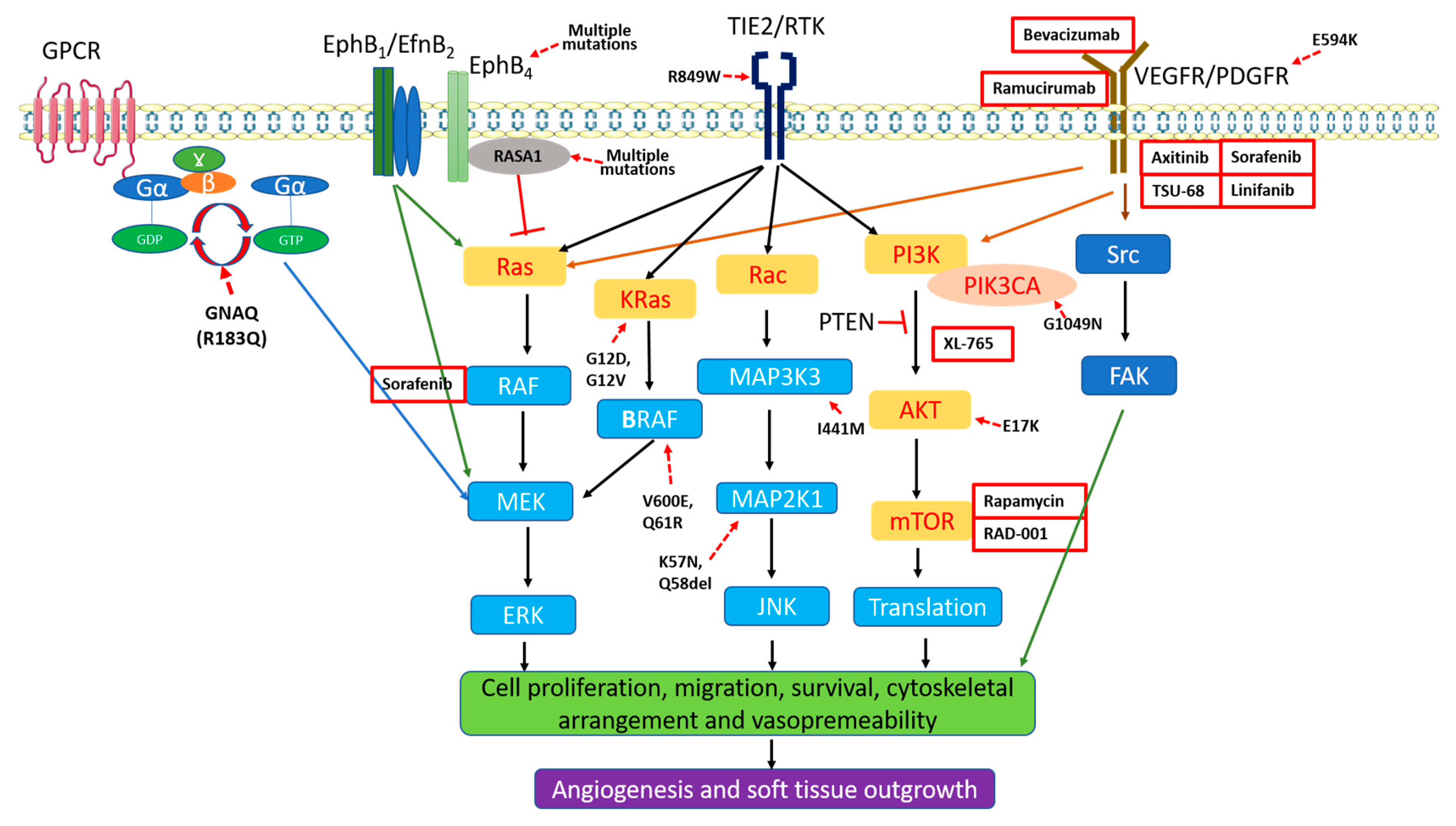
3.3. Aberrant Activation of MAPK and PI3K in PWS Vasculatures
4. Pathogenesis of PWS/SWS
4.1. Nerve Defect
4.2. Genetic Mutations
4.2.1. RASA1
4.2.2. GNAQ Mutation
4.2.3. PI3K and Other Mutations
5. Anti-Angiogenesis Therapies for Skin Lesions of SWS/PWS
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Tan, W.; Wang, J.; Zhou, F.; Gao, L.; Rong, Y.; Liu, H.; Sukanthanag, A.; Wang, G.; Mihm, M.C., Jr.; Chen, D.B.; et al. Coexistence of ephb1 and ephrinb2 in port wine stain endothelial progenitor cells contributes to clinicopathological vasculature dilatation. Br. J. Dermatol. 2017, 177, 1601–1611. [Google Scholar] [CrossRef]
- Geronemus, R.G.; Ashinoff, R. The medical necessity of evaluation and treatment of port-wine stains. J. Dermatol. Surg. Oncol. 1991, 17, 76–79. [Google Scholar] [CrossRef]
- Lever, W.F.; Schaumburg-Lever, G. Histopathology of the Skin, 7th ed.; J.B. Lippincott Co.: Philadelphia, PA, USA, 1990. [Google Scholar]
- Zallmann, M.; Mackay, M.T.; Leventer, R.J.; Ditchfield, M.; Bekhor, P.S.; Su, J.C. Retrospective review of screening for sturge-weber syndrome with brain magnetic resonance imaging and electroencephalography in infants with high-risk port-wine stains. Pediatr. Dermatol. 2018, 35, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, S.; Ercal, D. Genetic evaluation of common neurocutaneous syndromes. Pediatr. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Javaid, U.; Ali, M.H.; Jamal, S.; Butt, N.H. Pathophysiology, diagnosis, and management of glaucoma associated with sturge-weber syndrome. Int. Ophthalmol. 2018, 38, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Chernova, M.; Gao, L.; Sun, V.; Liu, H.; Jia, W.; Langer, S.; Wang, G.; Mihm, M.C., Jr.; Nelson, J.S. Sustained activation of c-jun n-terminal and extracellular signal regulated kinases in port wine stain blood vessels. J. Am. Acad. Dermatol. 2014, 71, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Gao, L.; Tan, W.; Guo, W.; Zhao, T.; Nelson, J.S.; Wang, G. Activation of pkcα and pi3k kinases in hypertrophic and nodular port wine stain lesions. Am. J. Dermatopathol. 2017, 39, 747–752. [Google Scholar] [CrossRef]
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge-weber syndrome and port-wine stains caused by somatic mutation in gnaq. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef]
- Lian, C.G.; Sholl, L.M.; Zakka, L.R.; O, T.M.; Liu, C.; Xu, S.; Stanek, E.; Garcia, E.; Jia, Y.; MacConaill, L.E.; et al. Novel genetic mutations in a sporadic port-wine stain. JAMA Dermatol. 2014, 150, 1336–1340. [Google Scholar] [CrossRef]
- Wassef, M.; Blei, F.; Adams, D.; Alomari, A.; Baselga, E.; Berenstein, A.; Burrows, P.; Frieden, I.J.; Garzon, M.C.; Lopez-Gutierrez, J.C.; et al. Vascular anomalies classification: Recommendations from the international society for the study of vascular anomalies. Pediatrics 2015, 136, e203–e214. [Google Scholar] [CrossRef]
- Comi, A. Current therapeutic options in sturge-weber syndrome. Semin. Pediatr. Neurol. 2015, 22, 295–301. [Google Scholar] [CrossRef]
- Mulliken, J.B.; Young, A.R. Vascular Birthmarks-Hemangiomas and Malformations; W.B. Saunders Co.: Philadelphia, PA, USA, 1988. [Google Scholar]
- Jacobs, A.H.; Walton, R.G. The incidence of birthmarks in the neonate. Pediatrics 1976, 58, 218–222. [Google Scholar] [CrossRef]
- Pratt, A.G. Birthmarks in infants. Arch. Dermatol. Syphilol. 1953, 67, 302–305. [Google Scholar] [CrossRef]
- Brightman, L.A.; Geronemus, R.G.; Reddy, K.K. Laser treatment of port-wine stains. Clin. Cosmet. Investig. Dermatol. 2015, 8, 27–33. [Google Scholar]
- Minkis, K.; Geronemus, R.G.; Hale, E.K. Port wine stain progression: A potential consequence of delayed and inadequate treatment? Lasers Surg. Med. 2009, 41, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.K.; Taber, S.F.; Ball, K.L.; Padwa, B.L.; Mulliken, J.B. Sturge-weber syndrome: Soft-tissue and skeletal overgrowth. J. Craniofac. Surg. 2009, 20 (Suppl. 1), 617–621. [Google Scholar] [CrossRef]
- Klapman, M.H.; Yao, J.F. Thickening and nodules in port-wine stains. J. Am. Acad. Dermatol. 2001, 44, 300–302. [Google Scholar] [CrossRef]
- Passeron, T.; Salhi, A.; Mazer, J.M.; Lavogiez, C.; Mazereeuw-Hautier, J.; Galliot, C.; Collet-Villette, A.M.; Labreze, C.; Boon, L.; Hardy, J.P.; et al. Prognosis and response to laser treatment of early-onset hypertrophic port-wine stains (PWS). J. Am. Acad. Dermatol. 2016, 75, 64–68. [Google Scholar] [CrossRef]
- Savas, J.A.; Ledon, J.A.; Franca, K.; Chacon, A.; Nouri, K. Pulsed dye laser-resistant port-wine stains: Mechanisms of resistance and implications for treatment. Br. J. Dermatol. 2013, 168, 941–953. [Google Scholar] [CrossRef]
- Lee, J.W.; Chung, H.Y. Capillary malformations (port wine stains) of the head and neck: Natural history, investigations, laser, and surgical management. Otolaryngol. Clin. N. Am. 2018, 51, 197–211. [Google Scholar] [CrossRef]
- Mills, C.M.; Lanigan, S.W.; Hughes, J.; Anstey, A.V. Demographic study of port wine stain patients attending a laser clinic: Family history, prevalence of naevus anaemicus and results of prior treatment. Clin. Exp. Dermatol. 1997, 22, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Renfro, L.; Geronemus, R.G. Anatomical differences of port-wine stains in response to treatment with the pulsed dye laser. Arch. Dermatol. 1993, 129, 182–188. [Google Scholar] [CrossRef]
- Mehta, M.; Salas, A.H.; Fay, A. Trigeminal dermatome distribution in patients with glaucoma and facial port wine stain. Dermatology 2009, 219, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chung, H.Y.; Cerrati, E.W.; O, T.M.; Waner, M. The natural history of soft tissue hypertrophy, bony hypertrophy, and nodule formation in patients with untreated head and neck capillary malformations. Dermatol. Surg. 2015, 41, 1241–1245. [Google Scholar] [CrossRef]
- Kalick, S.M. Toward an interdisciplinary psychology of appearances. Psychiatry 1978, 41, 243–253. [Google Scholar] [CrossRef]
- Heller, A.; Rafman, S.; Zvagulis, I.; Pless, I.B. Birth-defects and psychosocial adjustment. Am. J. Dis. Child. 1985, 139, 257–263. [Google Scholar] [CrossRef]
- Malm, M.; Carlberg, M. Port-wine stain—A surgical and psychological problem. Ann. Plast. Surg. 1988, 20, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Eivazi, B.; Roessler, M.; Pfutzner, W.; Teymoortash, A.; Werner, J.A.; Happle, R. Port-wine stains are more than skin-deep! Expanding the spectrum of extracutaneous manifestations of nevi flammei of the head and neck. Eur. J. Dermatol. 2012, 22, 246–251. [Google Scholar]
- Swerlick, R.A.; Cooper, P.H. Pyogenic granuloma (lobular capillary hemangioma) within port-wine stains. J. Am. Acad. Dermatol. 1983, 8, 627–630. [Google Scholar] [CrossRef]
- Fonder, M.A.; Mamelak, A.J.; Kazin, R.A.; Cohen, B.A. Port-wine-stain-associated dermatitis: Implications for cutaneous vascular laser therapy. Pediatr. Dermatol. 2007, 24, 376–379. [Google Scholar] [CrossRef] [PubMed]
- ISSVA. Issva classification of vascular anomalies. 2014. [Google Scholar]
- Haggstrom, A.N.; Lammer, E.J.; Schneider, R.A.; Marcucio, R.; Frieden, I.J. Patterns of infantile hemangiomas: New clues to hemangioma pathogenesis and embryonic facial development. Pediatrics 2006, 117, 698–703. [Google Scholar] [CrossRef]
- Yu, Y.; Flint, A.F.; Mulliken, J.B.; Wu, J.K.; Bischoff, J. Endothelial progenitor cells in infantile hemangioma. Blood 2004, 103, 1373–1375. [Google Scholar] [CrossRef]
- Singh, A.K.; Keenaghan, M. Sturge-weber syndrome. 2019. [Google Scholar]
- Gao, L.; Yin, R.; Wang, H.; Guo, W.; Song, W.; Nelson, J.S.; Tan, W.; Wang, G. Ultrastructural characterization of hyperactive endothelial cells, pericytes and fibroblasts in hypertrophic and nodular port wine stain lesions. Br. J. Dermatol. 2017. [Google Scholar] [CrossRef]
- Tan, W.; Zakka, L.R.; Gao, L.; Wang, J.; Zhou, F.; Selig, M.K.; Anvari, R.; Sukanthanag, A.; Wang, G.; Mihm, M.C., Jr.; et al. Pathological alterations involve the entire skin physiological milieu in infantile and early-childhood port-wine stain. Br. J. Dermatol. 2017, 177, 293–296. [Google Scholar] [CrossRef]
- Yin, R.; Rice, S.J.; Wang, J.; Gao, L.; Tsai, J.; Anvari, R.T.; Zhou, F.; Liu, X.; Wang, G.; Tang, Y.; et al. Membrane trafficking and exocytosis are upregulated in port wine stain blood vessels. Histol. Histopathol. 2018, 18051. [Google Scholar]
- Breathnach, A.S. The Ultrastructure of Human Skin; Longman Group Limited: London, UK, 1971; p. 98. [Google Scholar]
- Neumuller, J.; Neumuller-Guber, S.E.; Lipovac, M.; Mosgoeller, W.; Vetterlein, M.; Pavelka, M.; Huber, J. Immunological and ultrastructural characterization of endothelial cell cultures differentiated from human cord blood derived endothelial progenitor cells. Histochem. Cell Biol. 2006, 126, 649–664. [Google Scholar] [CrossRef]
- Schneider, B.V.; Mitsuhashi, Y.; Schnyder, U.W. Ultrastructural observations in port wine stains. Arch. Dermatol. Res. 1988, 280, 338–345. [Google Scholar] [CrossRef]
- Li, W.; Mukouyama, Y.S. Tissue-specific venous expression of the eph family receptor ephb1 in the skin vasculature. Dev. Dyn. 2013, 242, 976–988. [Google Scholar] [CrossRef]
- Perera, P.; Kurban, A.K.; Ryan, T.J. The development of the cutaneous microvascular system in the newborn. Br. J. Dermtol. 1970, 82, 86–91. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 2005, 6, 462–475. [Google Scholar] [CrossRef]
- Wang, H.U.; Chen, Z.F.; Anderson, D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-b2 and its receptor eph-b4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef]
- Morrison, D.K. Map kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. Mapk signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. Pi3k/akt/mtor pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Vural, E.; Ramakrishnan, J.; Cetin, N.; Buckmiller, L.; Suen, J.Y.; Fan, C.Y. The expression of vascular endothelial growth factor and its receptors in port-wine stains. Otolaryngol. Head Neck Surg. 2008, 139, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Rydh, M.; Malm, M.; Jernbeck, J.; Dalsgaard, C.J. Ectatic blood vessels in port-wine stains lack innervation: Possible role in pathogenesis. Plast. Reconstr. Surg. 1991, 87, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Selim, M.M.; Kelly, K.M.; Nelson, J.S.; Wendelschafer-Crabb, G.; Kennedy, W.R.; Zelickson, B.D. Confocal microscopy study of nerves and blood vessels in untreated and treated port wine stains: Preliminary observations. Dermatol. Surg. 2004, 30, 892–897. [Google Scholar]
- Tallman, B.; Tan, O.T.; Morelli, J.G.; Piepenbrink, J.; Stafford, T.J.; Trainor, S.; Weston, W.L. Location of port-wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics 1991, 87, 323–327. [Google Scholar]
- Frigerio, A.; Wright, K.; Wooderchak-Donahue, W.; Tan, O.T.; Margraf, R.; Stevenson, D.A.; Grimmer, J.F.; Bayrak-Toydemir, P. Genetic variants associated with port-wine stains. PLoS ONE 2015, 10, e0133158. [Google Scholar] [CrossRef]
- Nakashima, M.; Miyajima, M.; Sugano, H.; Iimura, Y.; Kato, M.; Tsurusaki, Y.; Miyake, N.; Saitsu, H.; Arai, H.; Matsumoto, N. The somatic gnaq mutation c.548g>a (p.R183q) is consistently found in sturge-weber syndrome. J. Hum. Genet. 2014, 59, 691–693. [Google Scholar] [CrossRef]
- Couto, J.A.; Huang, L.; Vivero, M.P.; Kamitaki, N.; Maclellan, R.A.; Mulliken, J.B.; Bischoff, J.; Warman, M.L.; Greene, A.K. Endothelial cells from capillary malformations are enriched for somatic gnaq mutations. Plast. Reconstr. Surg. 2016, 137, 77e–82e. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Couto, J.A.; Pinto, A.; Alexandrescu, S.; Madsen, J.R.; Greene, A.K.; Sahin, M.; Bischoff, J. Somatic gnaq mutation is enriched in brain endothelial cells in sturge-weber syndrome. Pediatr. Neurol. 2017, 67, 59–63. [Google Scholar] [CrossRef]
- Tan, W.; Nadora, D.M.; Gao, L.; Wang, G.; Mihm, M.C., Jr.; Nelson, J.S. The somatic gnaq mutation (r183q) is primarily located within the blood vessels of port wine stains. J. Am. Acad. Dermatol. 2016, 74, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Eerola, I.; Boon, L.M.; Mulliken, J.B.; Burrows, P.E.; Dompmartin, A.; Watanabe, S.; Vanwijck, R.; Vikkula, M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by rasa1 mutations. Am. J. Hum. Genet. 2003, 73, 1240–1249. [Google Scholar] [CrossRef]
- Eerola, I.; Boon, L.M.; Watanabe, S.; Grynberg, H.; Mulliken, J.B.; Vikkula, M. Locus for susceptibility for familial capillary malformation (‘port-wine stain’) maps to 5q. Eur. J. Hum. Genet. 2002, 10, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Revencu, N.; Boon, L.M.; Mendola, A.; Cordisco, M.R.; Dubois, J.; Clapuyt, P.; Hammer, F.; Amor, D.J.; Irvine, A.D.; Baselga, E.; et al. Rasa1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum. Mutat. 2013, 34, 1632–1641. [Google Scholar] [CrossRef]
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic ras/mapk variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 2018, 128, 1496–1508. [Google Scholar] [CrossRef]
- Couto, J.A.; Vivero, M.P.; Kozakewich, H.P.; Taghinia, A.H.; Mulliken, J.B.; Warman, M.L.; Greene, A.K. A somatic map3k3 mutation is associated with verrucous venous malformation. Am. J. Hum. Genet. 2015, 96, 480–486. [Google Scholar] [CrossRef]
- Limaye, N.; Wouters, V.; Uebelhoer, M.; Tuominen, M.; Wirkkala, R.; Mulliken, J.B.; Eklund, L.; Boon, L.M.; Vikkula, M. Somatic mutations in angiopoietin receptor gene tek cause solitary and multiple sporadic venous malformations. Nat. Genet. 2009, 41, 118–124. [Google Scholar] [CrossRef]
- Kurek, K.C.; Luks, V.L.; Ayturk, U.M.; Alomari, A.I.; Fishman, S.J.; Spencer, S.A.; Mulliken, J.B.; Bowen, M.E.; Yamamoto, G.L.; Kozakewich, H.P.; et al. Somatic mosaic activating mutations in pik3ca cause cloves syndrome. Am. J. Hum. Genet. 2012, 90, 1108–1115. [Google Scholar] [CrossRef]
- Amyere, M.; Revencu, N.; Helaers, R.; Pairet, E.; Baselga, E.; Cordisco, M.; Chung, W.; Dubois, J.; Lacour, J.P.; Martorell, L.; et al. Germline loss-of-function mutations in ephb4 cause a second form of capillary malformation-arteriovenous malformation (cm-avm2) deregulating ras-mapk signaling. Circulation 2017, 136, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Vikkula, M.; Boon, L.M.; Carraway, K.L.; Calvert, J.T.; Diamonti, A.J.; Goumnerov, B.; Pasyk, K.A.; Marchuk, D.A.; Warman, M.L.; Cantley, L.C.; et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase tie2. Cell 1996, 87, 1181–1190. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Sapp, J.C.; Teer, J.K.; Johnston, J.J.; Finn, E.M.; Peters, K.; Turner, J.; Cannons, J.L.; Bick, D.; Blakemore, L.; et al. A mosaic activating mutation in akt1 associated with the proteus syndrome. N. Engl. J. Med. 2011, 365, 611–619. [Google Scholar] [CrossRef]
- Thomas, A.C.; Zeng, Z.; Riviere, J.B.; O’Shaughnessy, R.; Al-Olabi, L.; St-Onge, J.; Atherton, D.J.; Aubert, H.; Bagazgoitia, L.; Barbarot, S.; et al. Mosaic activating mutations in gna11 and gnaq are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J. Investig. Dermatol. 2016, 136, 770–778. [Google Scholar] [CrossRef]
- Smoller, B.R.; Rosen, S. Port-wine stains. A disease of altered neural modulation of blood vessels? Arch. Dermatol. 1986, 122, 177–179. [Google Scholar] [CrossRef]
- Rosen, S.; Smoller, B. Pathogenesis of port wine stains. A new hypothesis. Med. Hypotheses 1987, 22, 365–368. [Google Scholar] [CrossRef]
- Hershkovitz, D.; Bercovich, D.; Sprecher, E.; Lapidot, M. Rasa1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br. J. Dermatol. 2008, 158, 1035–1040. [Google Scholar] [CrossRef]
- Cai, R.; Liu, F.; Hua, C.; Yu, Z.; Ramien, M.; Malic, C.; Yu, W.; Zhang, X.; Liu, Y.; Jin, Y.; et al. A novel rasa1 mutation causing capillary malformation-arteriovenous malformation (cm-avm): The first genetic clinical report in east asia. Hereditas 2018, 155, 24. [Google Scholar] [CrossRef]
- Pamonsinlapatham, P.; Hadj-Slimane, R.; Lepelletier, Y.; Allain, B.; Toccafondi, M.; Garbay, C.; Raynaud, F. P120-ras gtpase activating protein (rasgap): A multi-interacting protein in downstream signaling. Biochimie 2009, 91, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Henkemeyer, M.; Rossi, D.J.; Holmyard, D.P.; Puri, M.C.; Mbamalu, G.; Harpal, K.; Shih, T.S.; Jacks, T.; Pawson, T. Vascular system defects and neuronal apoptosis in mice lacking ras gtpase-activating protein. Nature 1995, 377, 695–701. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Carvajal, R.D. Gnaq and gna11 mutations in uveal melanoma. Melanoma Res. 2014, 24, 525–534. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of gnaq in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Populo, H.; Vinagre, J.; Lopes, J.M.; Soares, P. Analysis of gnaq mutations, proliferation and mapk pathway activation in uveal melanomas. Br. J. Ophthalmol. 2011, 95, 715–719. [Google Scholar] [CrossRef]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant gq/11 promote uveal melanoma tumorigenesis by activating yap. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Martins, L.; Giovani, P.A.; Reboucas, P.D.; Brasil, D.M.; Haiter Neto, F.; Coletta, R.D.; Machado, R.A.; Puppin-Rontani, R.M.; Nociti, F.H., Jr.; Kantovitz, K.R. Computational analysis for gnaq mutations: New insights on the molecular etiology of sturge-weber syndrome. J. Mol. Graph. Model. 2017, 76, 429–440. [Google Scholar] [CrossRef]
- di Blasio, L.; Puliafito, A.; Gagliardi, P.A.; Comunanza, V.; Somale, D.; Chiaverina, G.; Bussolino, F.; Primo, L. Pi3k/mtor inhibition promotes the regression of experimental vascular malformations driven by pik3ca-activating mutations. Cell Death Dis. 2018, 9, 45. [Google Scholar] [CrossRef]
- Couto, J.A.; Huang, A.Y.; Konczyk, D.J.; Goss, J.A.; Fishman, S.J.; Mulliken, J.B.; Warman, M.L.; Greene, A.K. Somatic map2k1 mutations are associated with extracranial arteriovenous malformation. Am. J. Hum. Genet. 2017, 100, 546–554. [Google Scholar] [CrossRef]
- Anderson, R.R.; Parrish, J.A. Selective photothermolysis-precise microsurgery by selective absorption of pulsed radiation. Science 1983, 220, 524–527. [Google Scholar] [CrossRef]
- Nelson, J.S.; Milner, T.E.; Anvari, B.; Tanenbaum, B.S.; Kimel, S.; Svaasand, L.O.; Jacques, S.L. Dynamic epidermal cooling during pulsed laser treatment of port-wine stain: A new methodology with preliminary clinical evaluation. Arch. Dermatol. 1995, 131, 695–700. [Google Scholar] [CrossRef]
- Chang, C.J.; Nelson, J.S. Cryogen spray cooling and higher fluence pulsed dye laser treatment improve port-wine stain clearance while minimizing epidermal damage. Dermatol. Surg. 1999, 25, 767–772. [Google Scholar] [CrossRef]
- Nelson, J.S.; Milner, T.E.; Anvari, B.; Tanenbaum, B.S.; Svaasand, L.O.; Kimel, S. Dynamic epidermal cooling in conjunction with laser-induced photothermolysis of port wine stain blood vessels. Lasers Surg. Med. 1996, 19, 224–229. [Google Scholar] [CrossRef]
- Chang, C.J.; Kelly, K.M.; van Gemert, M.J.C.; Nelson, J.S. Comparing the effectiveness of 585-nm vs. 595-nm wavelength pulsed dye laser treatment of port wine stains in conjunction with cryogen spray cooling. Lasers Surg. Med. 2002, 31, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.H.; Chan, H.H.L.; Ho, W.S.; Yeung, C.K.; Nelson, J.S. Prospective study of pulsed dye laser in conjunction with cryogen spray cooling for treatment of port wine stains in chinese patients. Dermatol. Surg. 2003, 29, 909–915. [Google Scholar]
- Waldorf, H.A.; Alster, T.S.; McMillan, K.; Kauvar, A.N.B.; Geronemus, R.G.; Nelson, J.S. Effect of dynamic cooling on 585-nm pulsed dye laser treatment of port-wine stain birthmarks. Dermatol. Surg. 1997, 23, 657–662. [Google Scholar] [CrossRef]
- Fiskerstrand, E.J.; Ryggen, K.; Norvang, L.T.; Svaasand, L.O. Clinical effects of dynamic cooling during pulsed laser treatment of port-wine stains. Lasers Med. Sci. 1997, 12, 320–327. [Google Scholar] [CrossRef]
- Wen, X.; Li, Y.; Hamblin, M.R. Photodynamic therapy in dermatology beyond non-melanoma cancer: An update. Photodiagnosis Photodyn. Ther. 2017, 19, 140–152. [Google Scholar] [CrossRef]
- van der Horst, C.M.A.M.; Koster, P.H.L.; de Borgie, C.A.J.M.; Bossuyt, P.M.M.; van Gemert, M.J.C. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed dye-laser. N. Engl. J. Med. 1998, 338, 1028–1033. [Google Scholar] [CrossRef]
- Yohn, J.J.; Huff, J.C.; Aeling, J.L.; Walsh, P.; Morelli, J.G. Lesion size is a factor for determining the rate of port-wine stain clearing following pulsed dye laser treatment in adults. Cutis 1997, 59, 267–270. [Google Scholar]
- Katugampola, G.A.; Lanigan, S.W. Five years’ experience of treating port wine stains with the flashlamp-pumped pulsed dye laser. Br. J. Dermatol. 1997, 137, 750–754. [Google Scholar] [CrossRef]
- Huikeshoven, M.; Koster, P.H.L.; de Borgie, C.; Beek, J.F.; van Gemert, M.J.C.; van der Horst, C. Redarkening of port-wine stains 10 years after pulsed-dye-laser treatment. N. Engl. J. Med. 2007, 356, 1235–1240. [Google Scholar] [CrossRef]
- Phung, T.L.; Oble, D.A.; Jia, W.; Benjamin, L.E.; Mihm, M.C., Jr.; Nelson, J.S. Can the wound healing response of human skin be modulated after laser treatment and the effects of exposure extended? Implications on the combined use of the pulsed dye laser and a topical angiogenesis inhibitor for treatment of port wine stain birthmarks. Lasers Surg. Med. 2008, 40, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Fiskerstrand, E.J.; Svaasand, L.O.; Kopstad, G.; Dalaker, M.; Norvang, L.T.; Volden, G. Laser treatment of port wine stains: Therapeutic outcome in relation to morphological parameters. Br. J. Dermatol. 1996, 134, 1039–1043. [Google Scholar] [CrossRef]
- Hohenleutner, U.; Hilbert, M.; Wlotzke, U.; Landthaler, M. Epidermal damage and limited coagulation depth with the flashlamp-pumped pulsed dye-laser—A histochemical-study. J. Investig. Dermatol. 1995, 104, 798–802. [Google Scholar] [CrossRef]
- Jia, W.; Choi, B.; Franco, W.; Lotfi, J.; Majaron, B.; Aguilar, G.; Nelson, J.S. Treatment of cutaneous vascular lesions using multiple-intermittent cryogen spurts and two-wavelength laser pulses: Numerical and animal studies. Lasers Surg. Med. 2007, 39, 494–503. [Google Scholar] [CrossRef]
- Coulon, C.; Georgiadou, M.; Roncal, C.; De Bock, K.; Langenberg, T.; Carmeliet, P. From vessel sprouting to normalization: Role of the prolyl hydroxylase domain protein/hypoxia-inducible factor oxygen-sensing machinery. Aterioscler. Thromb. Vasc. Biol. 2010, 30, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vegf-a: A critical regulator of blood vessel growth. Eur. Cytokine Netw. 2009, 20, 158–163. [Google Scholar] [PubMed]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. Vegf-a and the induction of pathological angiogenesis. Annu. Rev. Pathol. 2007, 2, 251–275. [Google Scholar] [CrossRef] [PubMed]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Kwon, Y.S.; Hong, H.S.; Kim, J.C.; Shin, J.S.; Son, Y. Inhibitory effect of rapamycin on corneal neovascularization in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2005, 46, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Phan, S.; Nadora, D.M.; Chernova, M.; Sun, V.; Preciado, S.M.; Ballew, B.; Jia, Z.; Jia, W.; Wang, G.; et al. Topical rapamycin systematically suppresses the early stages of pulsed dye laser-induced angiogenesis pathways. Lasers Surg. Med. 2014, 46, 679–688. [Google Scholar] [CrossRef]
- Gao, L.; Nadora, D.M.; Phan, S.; Chernova, M.; Sun, V.; Preciado, S.M.; Jia, W.; Wang, G.; Mihm, M.C., Jr.; Nelson, J.S.; et al. Topical axitinib suppresses angiogenesis pathways induced by pulsed dye laser. Br. J. Dermatol. 2015, 172, 669–676. [Google Scholar] [CrossRef]
- Passeron, T.; Maza, A.; Fontas, E.; Toubel, G.; Vabres, P.; Livideanu, C.; Mazer, J.M.; Rossi, B.; Boukari, F.; Harmelin, Y.; et al. Treatment of port wine stains with pulsed dye laser and topical timolol: A multicenter randomized controlled trial. Br. J. Dermatol. 2014, 170, 1350–1353. [Google Scholar] [CrossRef]
- Tremaine, A.M.; Armstrong, J.; Huang, Y.C.; Elkeeb, L.; Ortiz, A.; Harris, R.; Choi, B.; Kelly, K.M. Enhanced port-wine stain lightening achieved with combined treatment of selective photothermolysis and imiquimod. J. Am. Acad. Dermatol. 2012, 66, 634–641. [Google Scholar] [CrossRef]
- Marques, L.; Nunez-Cordoba, J.M.; Aguado, L.; Pretel, M.; Boixeda, P.; Nagore, E.; Baselga, E.; Redondo, P. Topical rapamycin combined with pulsed dye laser in the treatment of capillary vascular malformations in sturge-weber syndrome: Phase ii, randomized, double-blind, intraindividual placebo-controlled clinical trial. J. Am. Acad. Dermatol. 2015, 72, 151–158. [Google Scholar] [CrossRef]
- Nelson, J.S.; Jia, W.; Phung, T.L.; Mihm, M.C., Jr. Observations on enhanced port wine stain blanching induced by combined pulsed dye laser and rapamycin administration. Lasers Surg. Med. 2011, 43, 939–942. [Google Scholar] [CrossRef]
- Musalem, H.M.; Alshaikh, A.A.; Tuleimat, L.M.; Alajlan, S. Outcome with topical sirolimus for port wine stain malformations after unsatisfactory results with pulse dye laser treatment alone. Ann. Saudi Med. 2018, 38, 376–380. [Google Scholar] [CrossRef]
- Griffin, T.D., Jr.; Foshee, J.P.; Finney, R.; Saedi, N. Port wine stain treated with a combination of pulsed dye laser and topical rapamycin ointment. Lasers Surg. Med. 2016, 48, 193–196. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Mutations | Mutation Frequency Ranges (%) | Average Mutation Frequency *(%) | Positive Rate in Patients | Diagnosis or Sample Resource | Refs |
|---|---|---|---|---|---|---|
| GNAQ | R183Q | 1.1–18 | 3.98 ± 3.84 | 23 of 26 | PWS/SWS | [9] |
| 1.73–7.42 | 3.86 ± 1.91 | 9 of 12 | PWS | [54] | ||
| 3.6–8.9 | 5.59 ± 1.82 | 12 of 15 | SWS | [55] | ||
| 1.9–11.1 | 5.56 ± 2.65 | 8 of 13 | PWS/SWS | [56] | ||
| 2.8–11.3 | 7.05 ± 6.01 | 2 # | PWS, skin EC & | |||
| 7.6–42.9 | 27.35 ± 17.75 | 4 # | SWS, brain EC & | |||
| 14.7–21.0 | 17.85 ± 4.45 | 2 of 2 | SWS, brain EC & | [57] | ||
| 3.16–12.38 | 7.85 ± 4.18 | 6 of 10 | PWS, skin BV & | [58] | ||
| 2.67–22.17 | 8.81 ± 7.64 | 4 of 10 | PWS, HG/CT & | |||
| RASA1 | RASA1c.475_476delCT, RASA1c.512delT, RASA1c.1579_1582delGTCT, RASA1c.2336_2337delGC, Q446X, and C540Y | n.a. | n.a. | 6 of 17 | Familial PWS-AVM | [59,60] |
| 58 distinct mutations | 68 out of 100 | [61] | ||||
| KRAS | G12D | 2–30 (skin) | 16.25 ± 15.33 | 4 of 160 | High/Low flow VM | [62] |
| G12V | 3–5 (skin) | 3.67 ± 1.15 | 3 of 160 | |||
| Q61H | 5 | 5 | 1 of 160 | |||
| MAP2K1 | K57N | 2–7 | 4.50 ± 3.53 | 2 of 160 | ||
| Q58_E62del | 4 | 1 of 160 | ||||
| F53_Q58del | 6 | 1 of 160 | ||||
| BRAF | V600E | 26 | 1 of 160 | |||
| Q61R | 7 | 1 of 160 | ||||
| MAP3K3 | I441M | 5.5–19.3 | 11.13 ± 5.52 | 6 of 10 | VVM | [63] |
| TEK | L914F | 4.66–48.32 | 20.34 ± 14.61 | 24 of 57 | hereditary mucocutaneous VM | [64] |
| Y897H, Y897S, Y897F, Y897C, R915C, R915L, S917I | 4.55–34.90 | 4 of 57 | ||||
| PIK3CA | E542K | 6–8 | 7.00 ± 1.41 | 2 # | GLOVES/PWS/AVM/LM/VM | [65] |
| C420R | 3–11 | 7.00 ± 5.65 | 2 # | |||
| G1049N | 5 | 1 # | PWS | [10] | ||
| SMARCA4 | E514Q | 7 | ||||
| EPHA3 | S456C | 7 | ||||
| MYB | G349R | 10 | ||||
| PDGFR-β | E594K | 6 | ||||
| EPHB4 | 47 distinct mutations | n.a. | 54 of 365 | PWS-AVM | [66] | |
| Tie2 | R849W | n.a. | 2 families | inherited VM | [67] | |
| AKT1 | E17K | 3.6–51 | 22.43 ± 16.77 | 26 of 29 | Proteus syndrome | [68] |
| GNA11 | R183C | 5.3–9.6 | 7.45 ± 3.04 | 2 of 8 | Vascular skin lesion of PPV | [69] |
| R183Q | 5.0–6.4 | 5.70 ± 0.99 | 2 of 8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, V.; Hochman, M.; Mihm, M.C., Jr.; Nelson, J.S.; Tan, W. The Pathogenesis of Port Wine Stain and Sturge Weber Syndrome: Complex Interactions between Genetic Alterations and Aberrant MAPK and PI3K Activation. Int. J. Mol. Sci. 2019, 20, 2243. https://doi.org/10.3390/ijms20092243
Nguyen V, Hochman M, Mihm MC Jr., Nelson JS, Tan W. The Pathogenesis of Port Wine Stain and Sturge Weber Syndrome: Complex Interactions between Genetic Alterations and Aberrant MAPK and PI3K Activation. International Journal of Molecular Sciences. 2019; 20(9):2243. https://doi.org/10.3390/ijms20092243
Chicago/Turabian StyleNguyen, Vi, Marcelo Hochman, Martin C. Mihm, Jr., J. Stuart Nelson, and Wenbin Tan. 2019. "The Pathogenesis of Port Wine Stain and Sturge Weber Syndrome: Complex Interactions between Genetic Alterations and Aberrant MAPK and PI3K Activation" International Journal of Molecular Sciences 20, no. 9: 2243. https://doi.org/10.3390/ijms20092243
APA StyleNguyen, V., Hochman, M., Mihm, M. C., Jr., Nelson, J. S., & Tan, W. (2019). The Pathogenesis of Port Wine Stain and Sturge Weber Syndrome: Complex Interactions between Genetic Alterations and Aberrant MAPK and PI3K Activation. International Journal of Molecular Sciences, 20(9), 2243. https://doi.org/10.3390/ijms20092243