On-Membrane Dynamic Interplay between Anti-GM1 IgG Antibodies and Complement Component C1q

 , , , , , , ,
, , , , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Epitope Mapping of GB2
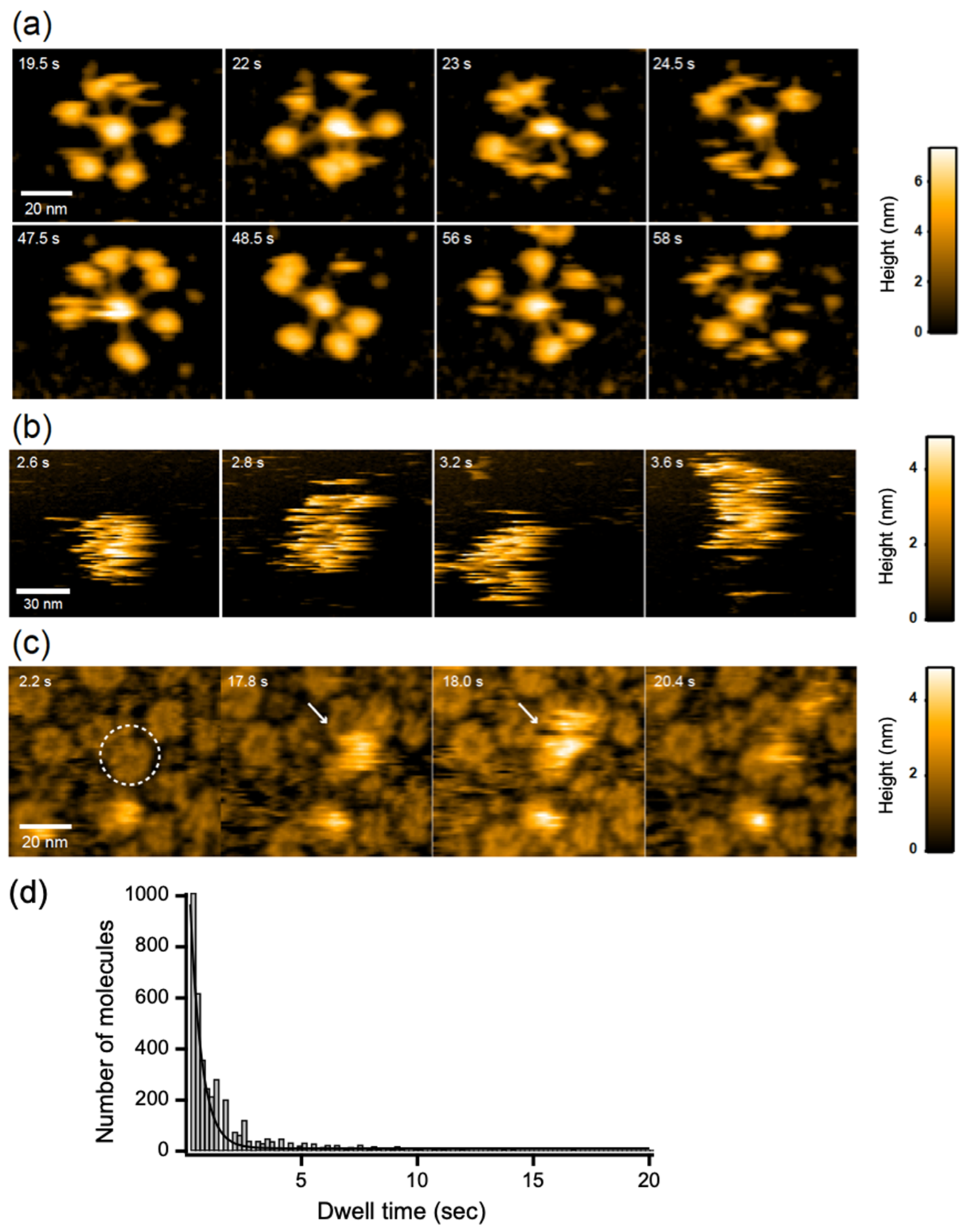
2.2. IgG Assembly on Antigen-Incorporated Membranes
2.3. C1q Interaction with the IgG Hexameric Ring Formed on Membranes
2.4. Effect of Protein A Binding and Disulfide Cleavage on IgG Hexameric Ring Formation
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Protein Preparation
4.1.1. Antibody
4.1.2. Protein A
4.1.3. C1q
4.1.4. Cholera Toxin B Subunit
4.3. NMR Spectroscopy
4.4. X-Ray Crystallography
4.5. PDB Accession Codes
4.6. HS-AFM
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IgG | immunoglobulin G |
| HS-AFM | high-speed atomic force microscopy |
| HSQC | heteronuclear single-quantum correlation |
| STD-NMR | saturation transfer difference nuclear magnetic resonance |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
References
- Delves, P.; Martin, S.; Burton, D.; Roitt, I. Roitt’s Essential Immunology, 13th ed.; WILEY-Blackwell: Hoboken, NJ, USA, 2016. [Google Scholar]
- Boyle, M.D.P. The type 1 bacterial immunoglobulin-binding protein: Staphylococcal protein A. Bact. Immunoglobulin-Bind. Proteins 1990, 1, 17–28. [Google Scholar]
- Bjorck, L.; Akerstorm, B. Streptococcal protein G. In Bacterial Immunoglobulin-Binding Proteins; Boyle, M.D.P., Ed.; Academic Press, Inc.: San Diego, CA, USA, 1990; Volume 1, pp. 113–126. [Google Scholar]
- Griffiss, J.M.; Schneider, H.; Mandrell, R.E.; Yamasaki, R.; Jarvis, G.A.; Kim, J.J.; Gibson, B.W.; Hamadeh, R.; Apicella, M.A. Lipooligosaccharides: The principal glycolipids of the neisserial outer membrane. Rev. Infect Dis. 1988, 10, S287–S295. [Google Scholar] [CrossRef]
- Schnaar, R.L.; Kinoshita, T. Glycosphingolipids. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanly, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2017; pp. 125–135. [Google Scholar]
- Yuki, N.; Susuki, K.; Koga, M.; Nishimoto, Y.; Odaka, M.; Hirata, K.; Taguchi, K.; Miyatake, T.; Furukawa, K.; Kobata, T.; et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barré syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 11404–11409. [Google Scholar] [CrossRef] [Green Version]
- Yuki, N.; Hartung, H.P. Guillain-Barré syndrome. N. Engl. J. Med. 2012, 366, 2294–2304. [Google Scholar] [CrossRef]
- Koga, M.; Yuki, N.; Hirata, K.; Morimatsu, M.; Mori, M.; Kuwabara, S. Anti-GM1 antibody IgG subclass: A clinical recovery predictor in Guillain-Barré syndrome. Neurology 2003, 60, 1514–1518. [Google Scholar] [CrossRef]
- Yogo, R.; Yamaguchi, Y.; Watanabe, H.; Yagi, H.; Satoh, T.; Nakanishi, M.; Onitsuka, M.; Omasa, T.; Shimada, M.; Maruno, T.; et al. The Fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Sci. Rep. 2019, 9, 11957. [Google Scholar] [CrossRef]
- Houliston, R.S.; Yuki, N.; Hirama, T.; Khieu, N.H.; Brisson, J.R.; Gilbert, M.; Jarrell, H.C. Recognition characteristics of monoclonal antibodies that are cross-reactive with gangliosides and lipooligosaccharide from Campylobacter jejuni strains associated with Guillain-Barré and Fisher syndromes. Biochemistry 2007, 46, 36–44. [Google Scholar] [CrossRef]
- Wang, G.; de Jong, R.N.; van den Bremer, E.T.; Beurskens, F.J.; Labrijn, A.F.; Ugurlar, D.; Gros, P.; Schuurman, J.; Parren, P.W.; Heck, A.J. Molecular basis of assembly and activation of complement component C1 in complex with immunoglobulin G1 and antigen. Mol. Cell 2016, 63, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef] [Green Version]
- Strasser, J.; de Jong, R.N.; Beurskens, F.J.; Wang, G.B.; Heck, A.J.R.; Schuurman, J.; Parren, P.W.H.I.; Hinterdorfer, P.; Preiner, J. Unraveling the macromolecular pathways of IgG oligomerization and complement activation on antigenic surfaces. Nano Lett. 2019, 19, 4787–4796. [Google Scholar] [CrossRef]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Nitsche-Schmitz, D.P.; Johansson, H.M.; Sastalla, I.; Reissmann, S.; Frick, I.M.; Chhatwal, G.S. Group G streptococcal igG binding molecules FOG and protein G have different impacts on opsonization by C1q. J. Biol. Chem. 2007, 282, 17530–17536. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Gouda, H.; Takaha, W.; Yoshino, A.; Matsunaga, C.; Arata, Y. 13C NMR study of the mode of interaction in solution of the B fragment of staphylococcal protein A and the Fc fragments of mouse immunoglobulin G. Febs Lett. 1993, 328, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-Å resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Deis, L.N.; Wu, Q.L.; Wang, Y.; Qi, Y.; Daniels, K.G.; Zhou, P.; Oas, T.G. Suppression of conformational heterogeneity at a protein-protein interface. Proc. Natl. Acad. Sci. USA 2015, 112, 9028–9033. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Lian, L.Y.; Barsukov, I.L.; Derrick, J.P.; Kim, H.H.; Tanaka, R.; Yoshino, A.; Shiraishi, M.; Shimada, I.; Arata, Y.; et al. Model for the complex between protein-G and an antibody Fc fragment in solution. Structure 1995, 3, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Sauer-Eriksson, A.E.; Kleywegt, G.J.; Uhlén, M.; Jones, T.A. Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG. Structure 1995, 3, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Brekke, O.H.; Michaelsen, T.E.; Sandlie, I. The structural requirements for complement activation by IgG - does it hinge on the hinge? Immunol. Today 1995, 16, 85–90. [Google Scholar] [CrossRef]
- Susuki, K.; Rasband, M.N.; Tohyama, K.; Koibuchi, K.; Okamoto, S.; Funakoshi, K.; Hirata, K.; Baba, H.; Yuki, N. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J. Neurosci. 2007, 27, 3956–3967. [Google Scholar] [CrossRef]
- Hafer-Macko, C.; Hsieh, S.T.; Li, C.Y.; Ho, T.W.; Sheikh, K.; Cornblath, D.R.; McKhann, G.M.; Asbury, A.K.; Griffin, J.W. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann. Neurol. 1996, 40, 635–644. [Google Scholar] [CrossRef]
- Kominami, H.; Kobayashi, K.; Ido, S.; Kimiya, H.; Yamada, H. Immunoactivity of self-assembled antibodies investigated by atomic force microscopy. RSC Adv. 2018, 8, 29378–29384. [Google Scholar] [CrossRef] [Green Version]
- Yanaka, S.; Yogo, R.; Inoue, R.; Sugiyama, M.; Itoh, S.G.; Okumura, H.; Miyanoiri, Y.; Yagi, H.; Satoh, T.; Yamaguchi, T.; et al. Dynamic views of the Fc region of immunoglobulin G provided by experimental and computational observations. Antibodies 2019, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Peschke, B.; Keller, C.W.; Weber, P.; Quast, I.; Lünemann, J.D. Fc-Galactosylation of human immunoglobulin gamma isotypes improves C1q binding and enhances complement-dependent cytotoxicity. Front Immunol. 2017, 8, 646. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Anthony, R.M.; Ravetch, J.V. Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc. Natl. Acad. Sci. USA 2007, 104, 8433–8437. [Google Scholar] [CrossRef] [Green Version]
- Quast, I.; Keller, C.W.; Maurer, M.A.; Giddens, J.P.; Tackenberg, B.; Wang, L.X.; Münz, C.; Nimmerjahn, F.; Dalakas, M.C.; Lünemann, J.D. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Investig. 2015, 125, 4160–4170. [Google Scholar] [CrossRef] [Green Version]
- Fuse, T.; Ando, H.; Imamura, A.; Sawada, N.; Ishida, H.; Kiso, M.; Ando, T.; Li, S.C.; Li, Y.T. Synthesis and enzymatic susceptibility of a series of novel GM2 analogs. Glycoconj. J. 2006, 23, 329–343. [Google Scholar] [CrossRef]
- Komori, T.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. Study on systematizing the synthesis of the a-series ganglioside glycans GT1a, GD1a, and GM1 using the newly developed N-Troc-protected GM3 and GalN intermediates. Carbohydr. Res. 2009, 344, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Matsunaga, C.; Igarashi, T.; Kim, H.; Odaka, A.; Shimada, I.; Arata, Y. Complete assignment of the methionyl carbonyl carbon resonances in switch variant anti-dansyl antibodies labeled with [1-13C]methionine. Biochemistry 1991, 30, 270–278. [Google Scholar] [CrossRef]
- Sawada, J.; Terao, T.; Itoh, S.; Maeda, M.; Tsuji, A.; Hosoda, H.; Nambara, T. Production and characterization of monoclonal antibodies to 17α-hydroxyprogesterone. J. Steroid Biochem. 1987, 28, 405–410. [Google Scholar]
- Yonemasu, K.; Stroud, R.M. Clq: Rapid purification method for preparation of monospecific antisera and for biochemical studies. J. Immunol. 1971, 106, 304–313. [Google Scholar]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Cryst. D Biol. Cryst. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Cryst. Sect. D Biol. Cryst. 2011, 67, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Cryst. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Kolenko, P.; Dohnálek, J.; Dusková, J.; Skálová, T.; Collard, R.; Hasek, J. New insights into intra- and intermolecular interactions of immunoglobulins: Crystal structure of mouse IgG2b-Fc at 2.1-Å resolution. Immunology 2009, 126, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. Sect. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. Sect. D Biol. Cryst. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. Sect. D Biol. Cryst. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: φ, ψ and Cβ Deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Ando, T.; Kodera, N.; Takai, E.; Maruyama, D.; Saito, K.; Toda, A. A high-speed atomic force microscope for studying biological macromolecules. Proc. Natl. Acad. Sci. USA 2001, 98, 12468–12472. [Google Scholar] [CrossRef] [Green Version]
- Uchihashi, T.; Kodera, N.; Ando, T. Guide to video recording of structure dynamics and dynamic processes of proteins by high-speed atomic force microscopy. Nat. Protoc. 2012, 7, 1193–1206. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanaka, S.; Yogo, R.; Watanabe, H.; Taniguchi, Y.; Satoh, T.; Komura, N.; Ando, H.; Yagi, H.; Yuki, N.; Uchihashi, T.; et al. On-Membrane Dynamic Interplay between Anti-GM1 IgG Antibodies and Complement Component C1q. Int. J. Mol. Sci. 2020, 21, 147. https://doi.org/10.3390/ijms21010147
Yanaka S, Yogo R, Watanabe H, Taniguchi Y, Satoh T, Komura N, Ando H, Yagi H, Yuki N, Uchihashi T, et al. On-Membrane Dynamic Interplay between Anti-GM1 IgG Antibodies and Complement Component C1q. International Journal of Molecular Sciences. 2020; 21(1):147. https://doi.org/10.3390/ijms21010147
Chicago/Turabian StyleYanaka, Saeko, Rina Yogo, Hiroki Watanabe, Yuki Taniguchi, Tadashi Satoh, Naoko Komura, Hiromune Ando, Hirokazu Yagi, Nobuhiro Yuki, Takayuki Uchihashi, and et al. 2020. "On-Membrane Dynamic Interplay between Anti-GM1 IgG Antibodies and Complement Component C1q" International Journal of Molecular Sciences 21, no. 1: 147. https://doi.org/10.3390/ijms21010147
APA StyleYanaka, S., Yogo, R., Watanabe, H., Taniguchi, Y., Satoh, T., Komura, N., Ando, H., Yagi, H., Yuki, N., Uchihashi, T., & Kato, K. (2020). On-Membrane Dynamic Interplay between Anti-GM1 IgG Antibodies and Complement Component C1q. International Journal of Molecular Sciences, 21(1), 147. https://doi.org/10.3390/ijms21010147