Evading the AAV Immune Response in Mucopolysaccharidoses

 , ,
, , 
Abstract
:1. Introduction
2. Anti-AAV Antibodies and the AAV Capsid
3. Transgene Product Immune Response
4. Evading the Anti-Capsid Immune Response
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| MPS | mucopolysaccharidoses |
| GAG | glycosaminoglycan |
| LSD | lysosomal storage sisorder |
| ERT | enzyme replacement therapy |
| HSCT | hematopoietic stem cell transplantation |
| IC | intracerebral |
| ICS | intracisternal |
| ICSF | intracerebrospinal fluid |
| IV | intravenous |
| CNS | central nervous system |
| IDUA | iduronidase |
| uGAG | urinary glycosaminoglycans |
| HS | heparan sulfate |
| CSF | cerebrospinal fluid |
| IDS | iduronate-2-sulfate |
| SGSH | N-sulfoglucosamine sulfohydrolase |
| DQ | developmental quotient |
| BSID | Bayley scale of infant development |
| NAGLU | N-acetyl-alpha-glucosaminidase |
| LV | lentivirus |
| RV | retrovirus |
| ITR | inverted terminal repeat |
| ORF | open reading frame |
| ZFN | zinc finger nuclease |
| Cryo-EM | cryogenic electron microscopy |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| MHC | major histocompatibility complex |
| LLPC | long-lived plasma cell |
| nAb | neutralizing antibody |
References
- Neufeld, E.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Diseases; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019. [Google Scholar] [CrossRef] [PubMed]
- Lavery, C.; Hendriksz, C.J.; Jones, S.A. Mortality in Patients with Sanfilippo Syndrome. Orphanet J. Rare Dis. 2017, 12, 167–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.A.; Almássy, Z.; Beck, M.; Burt, K.; Clarke, J.T.; Giugliani, R.; Hendriksz, C.; Kroepfl, T.; Lavery, L.; Lin, S.; et al. Mortality and Cause of Death in Mucopolysaccharidosis Type II—a Historical Review Based on Data from the Hunter Outcome Survey (HOS). J. Inherit. Metab. Dis. 2009, 32, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Lavery, C.; Hendriksz, C. Mortality in Patients with Morquio Syndrome A. JIMD Rep. 2015, 15, 59–66. [Google Scholar]
- Montaño, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Çoker, M.; Bartholomew, D.; et al. Clinical Course of Sly Syndrome (Mucopolysaccharidosis Type VII). J. Med Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef]
- Moore, D.; Connock, M.J.; Wraith, E.; Lavery, C. The Prevalence of and Survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie Syndromes in the UK. Orphanet J. Rare Dis. 2008, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- Valayannopoulos, V.; Nicely, H.; Harmatz, P.; Turbeville, S. Mucopolysaccharidosis VI. Orphanet J. Rare Dis. 2010, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Hendriksz, C.; Harmatz, P.; Beck, M.; Simon, J.; Wood, T.; Lachman, R.; Orii, T. A Review of the Clinical Presentation and Diagnosis of Mucopolysaccharidosis IVA. Mol. Genet. Metab. 2013, 108, S90–S91. [Google Scholar] [CrossRef] [Green Version]
- Bitencourt, F.H.d.; Vieira, T.A.; Steiner, C.E.; Neto, J.C.; Boy, R.; Schwartz, I.V.D. Medical Costs Related to Enzyme Replacement Therapy for Mucopolysaccharidosis Types I, II, and VI in Brazil: A Multicenter Study. Value Health Reg. Issues 2015, 8, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme Replacement Therapy: Efficacy and Limitations. Ital. J. Pediatrics 2018, 44, 120. [Google Scholar] [CrossRef]
- Donald, S.A.; McIntyre, C.; Byers, S. Therapies for Neurological Disease in the Mucopolysaccharidoses. Curr. Gene Ther. 2011, 11, 132–143. [Google Scholar]
- Hobbs, J.R.; Barrett, A.J.; Chambers, D.; James, D.C.O.; Hugh-Jones, K.; Byrom, N.; Henry, K.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of Clinical Features of Hurler’s Disease and Biochemical Improvement After Treatment by Bone-Marrow Transplantation. Lancet 1981, 318, 709–712. [Google Scholar] [CrossRef]
- Stapleton, M.; Hoshina, H.; Sawamoto, K.; Kubaski, F.; Mason, R.W.; Mackenzie, W.G.; Theroux, M.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; et al. Critical Review of Current MPS Guidelines and Management. Mol. Genet. Metab. 2019, 126, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Yabe, H.; Tanaka, A.; Chinen, Y.; Kato, S.; Sawamoto, K.; Yasuda, E.; Shintaku, H.; Suzuki, Y.; Orii, T.; Tomatsu, S. Hematopoietic Stem Cell Transplantation for Morquio A Syndrome. Mol. Genet. Metab. 2016, 117, 84–94. [Google Scholar] [CrossRef] [Green Version]
- REGENXBIO Reports Fourth Quarter and Full-Year 2019 Financial Results and Operational Highlights. PR Newswire 2020. Available online: https://www.prnewswire.com/news-releases/regenxbio-reports-fourth-quarter-and-full-year-2019-financial-results-and-operational-highlights-301011953.html (accessed on 10 January 2020).
- Sangamo Announces Interim Results of Phase 1/2 EMPOWERS Study Evaluating SB-318 Zinc Finger Nuclease (ZFN) in Vivo Genome Editing Demonstrating Increased Leukocyte IDUA Activity in Patients with MPS I. Contify Life Science News 2019. Available online: https://investor.sangamo.com/news-releases/news-release-details/sangamo-announces-interim-results-phase-12-champions-study (accessed on 10 January 2020).
- Gene Therapy with Modified Autologous Hematopoietic Stem Cells for the Treatment of Patients with Mucopolysaccharidosis Type I, Hurler Variant (TigetT10_MPSIH). Available online: https://clinicaltrials.gov/ct2/show/NCT03488394 (accessed on 10 January 2020).
- REGENXBIO Announces Interim Data from Phase I/II Trial of RGX-121 for the Treatment of Mucopolysaccharidosis Type II (MPS II). PR Newswire 2019. Available online: https://www.prnewswire.com/news-releases/regenxbio-announces-interim-data-from-phase-iii-trial-of-rgx-121-for-the-treatment-of-mucopolysaccharidosis-type-ii-mps-ii-300976867.html (accessed on 10 January 2020).
- Muenzer, J.; Prada, C.E.; Burton, B.; Lau, H.A.; Ficicioglu, C.; Foo, C.W.P.; Vaidya, S.A.; Whitley, C.B.; Harmatz, P. CHAMPIONS: A Phase 1/2 Clinical Trial with Dose Escalation of SB-913 ZFN-Mediated in Vivo Human Genome Editing for Treatment of MPS II (Hunter Syndrome). Mol. Genet. Metab. 2019, 126, S104. [Google Scholar] [CrossRef]
- Lopes, J. Gene Therapy ABO-102 Preserves Cognitive Development in Young Children with Sanfilippo Type A, Interim Data Show. Sanfilippo News 2019. Available online: https://sanfilipponews.com/2019/08/08/type-a-gene-therapy-abo-102-preserves-cognitive-development-young-children-interim-data/ (accessed on 12 January 2020).
- Gene Therapy with Modified Autologous Hematopoietic Stem Cells for Patients with Mucopolysaccharidosis Type IIIA. Available online: https://clinicaltrials.gov/ct2/show/NCT04201405?term=gene+therapy&cond=Mucopolysaccharidoses&draw=2&rank=1. (accessed on 12 January 2020).
- Study of AAVrh10-H.SGSH Gene Therapy in Patients with Mucopolysaccharidosis Type IIIA (MPS IIIA) (AAVance). Available online: http://clinicaltrials.gov/ct2/show/NCT03612869?term=gene+therapy&cond=Mucopolysaccharidoses&draw=2&rank=2. (accessed on 12 January 2020).
- EU Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2015-000359-26. (accessed on 12 January 2020).
- Gene Transfer Clinical Trial for Mucopolysaccharidosis (MPS) IIIB (MPSIIIB). Available online: https://clinicaltrials.gov/ct2/show/NCT03315182?term=gene+therapy&cond=Mucopolysaccharidoses&draw=8&rank=6. (accessed on 12 January 2020).
- Gene Therapy in Patients with Mucopolysaccharidosis Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03173521. (accessed on 12 January 2020).
- Fraldi, A.; Serafini, M.; Sorrentino, N.C.; Gentner, B.; Aiuti, A.; Bernardo, M.E. Gene Therapy for Mucopolysaccharidoses: In Vivo and Ex Vivo Approaches. Ital. J. Pediatrics 2018, 44, 130–154. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Wijburg, F.A. Therapy for the Mucopolysaccharidoses. Rheumatology 2011, 50, v49–v59. [Google Scholar] [CrossRef] [Green Version]
- Naso, M.; Tomkowicz, B.; Perry III, W.; Strohl, W. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Sawamoto, K.; Chen, H.; Alméciga-Díaz, C.J.; Mason, R.W.; Tomatsu, S. Gene Therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68. [Google Scholar] [CrossRef]
- Daya, S.; Berns, K. Gene Therapy using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Grieger, J.C.; Samulski, R.J. Packaging Capacity of Adeno-Associated Virus Serotypes: Impact of Larger Genomes on Infectivity and Postentry Steps. J. Virol. 2005, 79, 9933–9944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.J.; Lane, M.D.; Padron, E.; Gurda, B.; McKenna, R.; Kohlbrenner, E.; Aslanidi, G.; Byrne, B.; Muzyczka, N.; Zolotukhin, S.; et al. Structure of Adeno-Associated Virus Serotype 8, a Gene Therapy Vector. J. Virol. 2007, 81, 12260–12271. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel Adeno-Associated Viruses from Rhesus Monkeys as Vectors for Human Gene Therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foust, K.D.; Montgomery, C.L.; Nurre, E.; Chan, C.M.; Hernandez, A.; Kaspar, B.K. Intravascular AAV9 Preferentially Targets Neonatal Neurons and Adult Astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMattia, M.A.; Nam, H.J.; Van Vliet, K.; Mitchell, M.; Bennett, A.; Gurda, B.L.; McKenna, R.; Olson, N.H.; Sinkovits, R.S.; Potter, M.; et al. Structural Insight into the Unique Properties of Adeno-Associated Virus Serotype 9. J. Virol. 2012, 86, 6947–6958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful Transduction of Liver in Hemophilia by AAV-Factor IX and Limitations Imposed by the Host Immune Response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Ferla, R.; O’Malley, T.; Calcedo, R.; O’Donnell, P.; Wang, P.; Cotugno, G.; Claudiani, P.; Wilson, J.M.; Haskins, M.; Auricchio, A. Gene Therapy for Mucopolysaccharidosis Type VI is Effective in Cats without Pre-Existing Immunity to AAV8. Hum. Gene Ther. 2013, 24, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Hinderer, C.; Bell, P.; Louboutin, J.; Zhu, Y.; Yu, H.; Lin, G.; Choa, R.; Gurda, B.L.; Bagel, J.; O’Donnell, P.; et al. Neonatal Systemic AAV Induces Tolerance to CNS Gene Therapy in MPS I Dogs and Nonhuman Primates. Mol. Ther. 2015, 23, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Martino, A.T.; Markusic, D.M. Immune Response Mechanisms Against AAV Vectors in Animal Models. Mol. Ther. Methods Clin. Dev. 2020, 17, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum. Gene Ther. 2017, 28, 161–1074. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Morizono, H.; Wang, L.; McCarter, R.; He, J.; Jones, D.; Batshaw, M.L.; Wilson, J.M. Adeno-Associated Virus Antibody Profiles in Newborns, Children, and Adolescents. Clin. Vaccine Immunol. 2011, 18, 1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-Existing Anti–Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide Epidemiology of Neutralizing Antibodies to Adeno-Associated Viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, J.L.; O’Callaghan, M.; Rubin, H.; Seymour, A. Low Seroprevalence of Neutralizing Antibodies Targeting Two Clade F AAV in Humans. Hum. Gene Ther. Clin. Dev. 2018, 29, 6–67. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Meadows, A.S.; Pineda, R.J.; Kunkler, K.L.; Truxal, K.V.; McBride, K.L.; Flanigan, K.M.; McCarty, D.M. Differential Prevalence of Antibodies Against Adeno-Associated Virus in Healthy Children and Patients with Mucopolysaccharidosis III: Perspective for AAV-Mediated Gene Therapy. Hum. Gene Ther. Clin. Dev. 2017, 28, 187–196. [Google Scholar] [CrossRef]
- Liu, Q.; Huang, W.; Zhang, H.; Wang, Y.; Zhao, J.; Song, A.; Xie, H.; Zhao, C.; Gao, D.; Wang, Y. Neutralizing Antibodies Against AAV2, AAV5 and AAV8 in Healthy and HIV-1-Infected Subjects in China: Implications for Gene Therapy using AAV Vectors. Gene Ther. 2014, 21, 732–738. [Google Scholar] [CrossRef]
- Perocheau, D.P.; Cunningham, S.C.; Lee, J.; Antinao Diaz, J.; Waddington, S.N.; Gilmour, K.; Eaglestone, S.; Lisowski, L.; Thrasher, A.J.; Alexander, I.E.; et al. Age-Related Seroprevalence of Antibodies Against AAV-LK03 in a UK Population Cohort. Hum. Gene Ther. 2019, 30, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Mimuro, J.; Mizukami, H.; Shima, M.; Matsushita, T.; Taki, M.; Muto, S.; Higasa, S.; Sakai, M.; Ohmori, T.; Madoiwa, S.; et al. The Prevalence of Neutralizing Antibodies Against Adeno-associated Virus Capsids is Reduced in Young Japanese Individuals. J. Med Virol. 2014, 86, 1990–1997. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune Responses to AAV Vectors: Overcoming Barriers to Successful Gene Therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-Associated Viruses are Widely Disseminated in Human Tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.; Agbandje-Mckenna, M. Mapping the AAV Capsid Host Antibody Response Toward the Development of Second Generation Gene Delivery Vectors. Front. Immunol. 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immune Epitope Database. Available online: https://www.iedb.org/ (accessed on 7 February 2020).
- Fitzpatrick, Z.; Leborgne, C.; Barbon, E.; Masat, E.; Ronzitti, G.; van Wittenberghe, L.; Vignaud, A.; Collaud, F.; Charles, S.; Simon Sola, M.; et al. Influence of Pre-Existing Anti-Capsid Neutralizing and Binding Antibodies on AAV Vector Transduction. Mol. Ther. Methods Clin. Dev. 2018, 9, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, R.W. Complexity of Immune Responses to AAV Transgene Products–Example of Factor IX. Cell. Immunol. 2019, 342, 103658. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Mays, L.E.; Wilson, J.M. The Complex and Evolving Story of T Cell Activation to AAV Vector-Encoded Transgene Products. Mol. Ther. 2011, 19, 16–27. [Google Scholar] [CrossRef]
- Colella, P.; Sellier, P.; Costa Verdera, H.; Puzzo, F.; van Wittenberghe, L.; Guerchet, N.; Daniele, N.; Gjata, B.; Marmier, S.; Charles, S.; et al. AAV Gene Transfer with Tandem Promoter Design Prevents Anti-Transgene Immunity and Provides Persistent Efficacy in Neonate Pompe Mice. Mol. Ther. Methods Clin. Dev. 2019, 12, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Cao, O.; Hoffman, B.E.; Moghimi, B.; Nayak, S.; Cooper, M.; Zhou, S.; Ertl, H.C.; High, K.A.; Herzog, R.W. Impact of the Underlying Mutation and the Route of Vector Administration on Immune Responses to Factor IX in Gene Therapy for Hemophilia B. Mol. Ther. 2009, 17, 1733–1742. [Google Scholar] [CrossRef]
- Mingozzi, F.; Liu, Y.; Dobrzynski, E.; Kaufhold, A.; Liu, J.H.; Wang, Y.; Arruda, V.R.; High, K.A.; Herzog, R.W. Induction of Immune Tolerance to Coagulation Factor IX Antigen by in Vivo Hepatic Gene Transfer. J. Clin. Investig. 2003, 111, 1347–1356. [Google Scholar] [CrossRef]
- Loduca, P.A.; Hoffman, B.E.; Herzog, R.W. Hepatic Gene Transfer as a Means of Tolerance Induction to Transgene Products. Curr. Gene Ther. 2009, 9, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Pastore, L.; Morral, N.; Zhou, H.; Garcia, R.; Parks, R.J.; Kochanek, S.; Graham, F.L.; Lee, B.; Beaudet, A.L. Use of a Liver-Specific Promoter Reduces Immune Response to the Transgene in Adenoviral Vectors. Hum. Gene Ther. 1999, 11, 1773–1781. [Google Scholar] [CrossRef]
- Knolle, P.A.; Gerken, G. Local Control of the Immune Response in the Liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Tian, Z. Liver-Mediated Adaptive Immune Tolerance. Front. Immunol. 2019, 10, 2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacParland, S.A.; Liu, J.C.; Ma, X.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single Cell RNA Sequencing of Human Liver Reveals Distinct Intrahepatic Macrophage Populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessitore, A.; Faella, A.; Parisi, F.; Haskins, M.; Auricchio, A. AAV-Mediated Gene Transfer to Muscle and Liver of MPS VI Animal Models. Mol. Ther. 2006, 13, S79. [Google Scholar] [CrossRef]
- Maheshri, N.; Kaspar, B.K.; Schaffer, D.V.; Koerber, J.T. Directed Evolution of Adeno-Associated Virus Yields Enhanced Gene Delivery Vectors. Nat. Biotechnol. 2006, 24, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Selot, R.; Arumugam, S.; Mary, B.; Cheemadan, S.; Jayandharan, G.R. Optimized AAV Rh.10 Vectors that Partially Evade Neutralizing Antibodies during Hepatic Gene Transfer. Front. Pharmacol. 2017, 8, 441. [Google Scholar] [CrossRef]
- Le, H.T.; Yu, Q.; Wilson, J.M.; Croyle, M.A. Utility of PEGylated Recombinant Adeno-Associated Viruses for Gene Transfer. J. Control. Release 2005, 108, 161–177. [Google Scholar] [CrossRef]
- Wang, D.; Li, S.; Li, J.; Gessler, D.J.; Xie, J.; Zhong, L.; Tran, K.; Van Vliet, K.; Ren, L.; Su, Q.; et al. A Rationally Engineered Capsid Variant of AAV9 for Systemic CNS-Directed and Peripheral Tissue-Detargeted Gene Delivery in Neonates. Mol. Ther. Methods Clin. Dev. 2018, 9, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Tse, L.V.; Klinc, K.A.; Madigan, V.J.; Castellanos Rivera, R.M.; Wells, L.F.; Havlik, L.P.; Smith, J.K.; Agbandje-McKenna, M.; Asokan, A. Structure-Guided Evolution of Antigenically Distinct Adeno-Associated Virus Variants for Immune Evasion. Proc. Natl. Acad. Sci. USA 2017, 114, E4812–E4821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asokan, A.; Schaffer, D.V.; Jude Samulski, R. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.J., III. An Autoradiographic Study of Plasma Cell and Lymphocyte Survival in Rat Popliteal Lymph Nodes. J. Immunol. 1964, 92, 673–681. [Google Scholar]
- Lykken, E.; Shyng, C.; Edwards, R.; Rozenberg, A.; Gray, S. Recent Progress and Considerations for AAV Gene Therapies Targeting the Central Nervous System. J. Neurodev. Disord. 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral Immunity due to Long-Lived Plasma Cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Holt, P.G.; Sedgwick, J.D.; O’Leary, C.; Krska, K.; Leivers, S. Long-Lived IgE- and IgG-Secreting Cells in Rodents Manifesting Persistent Antibody Responses. Cell. Immunol. 1984, 89, 281–289. [Google Scholar] [CrossRef]
- Velazquez, V.M.; Meadows, A.S.; Pineda, R.J.; Camboni, M.; McCarty, D.M.; Fu, H. Effective Depletion of Pre-Existing Anti-AAV Antibodies Requires Broad Immune Targeting. Mol. Ther. Methods Clin. Dev. 2017, 4, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [Green Version]
- Monteilhet, V.; Saheb, S.; Boutin, S.; Leborgne, C.; Veron, P.; Montus, M.; Moullier, P.; Benveniste, O.; Masurier, C. A 10 Patient Case Report on the Impact of Plasmapheresis upon Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 6, and 8. Mol. Ther. 2011, 19, 2084–2091. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-Associated AAV Vector as a Novel Gene Delivery System. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meliani, A.; Boisgerault, F.; Fitzpatrick, Z.; Marmier, S.; Leborgne, C.; Collaud, F.; Simon Sola, M.; Charles, S.; Ronzitti, G.; Vignaud, A.; et al. Enhanced Liver Gene Transfer and Evasion of Preexisting Humoral Immunity with Exosome-Enveloped AAV Vectors. Blood Adv. 2017, 1, 2019–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, L.T.; Lemus-Diaz, N.; Rinaldi Ferreira, R.; Böker, K.O.; Gruber, J. Enhanced Production of Exosome-Associated AAV by Overexpression of the Tetraspanin CD9. Mol. Ther. Methods Clin. Dev. 2018, 9, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orefice, N.S.; Souchet, B.; Braudeau, J.; Alves, S.; Piguet, F.; Collaud, F.; Ronzitti, G.; Tada, S.; Hantraye, P.; Mingozzi, F.; et al. Real-Time Monitoring of Exosome Enveloped-AAV Spreading by Endomicroscopy Approach: A New Tool for Gene Delivery in the Brain. Mol. Ther. Methods Clin. Dev. 2019, 14, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming Preexisting Humoral Immunity to AAV using Capsid Decoys. Sci. Transl. Med. 2013, 5, 194ra92. [Google Scholar] [CrossRef] [Green Version]


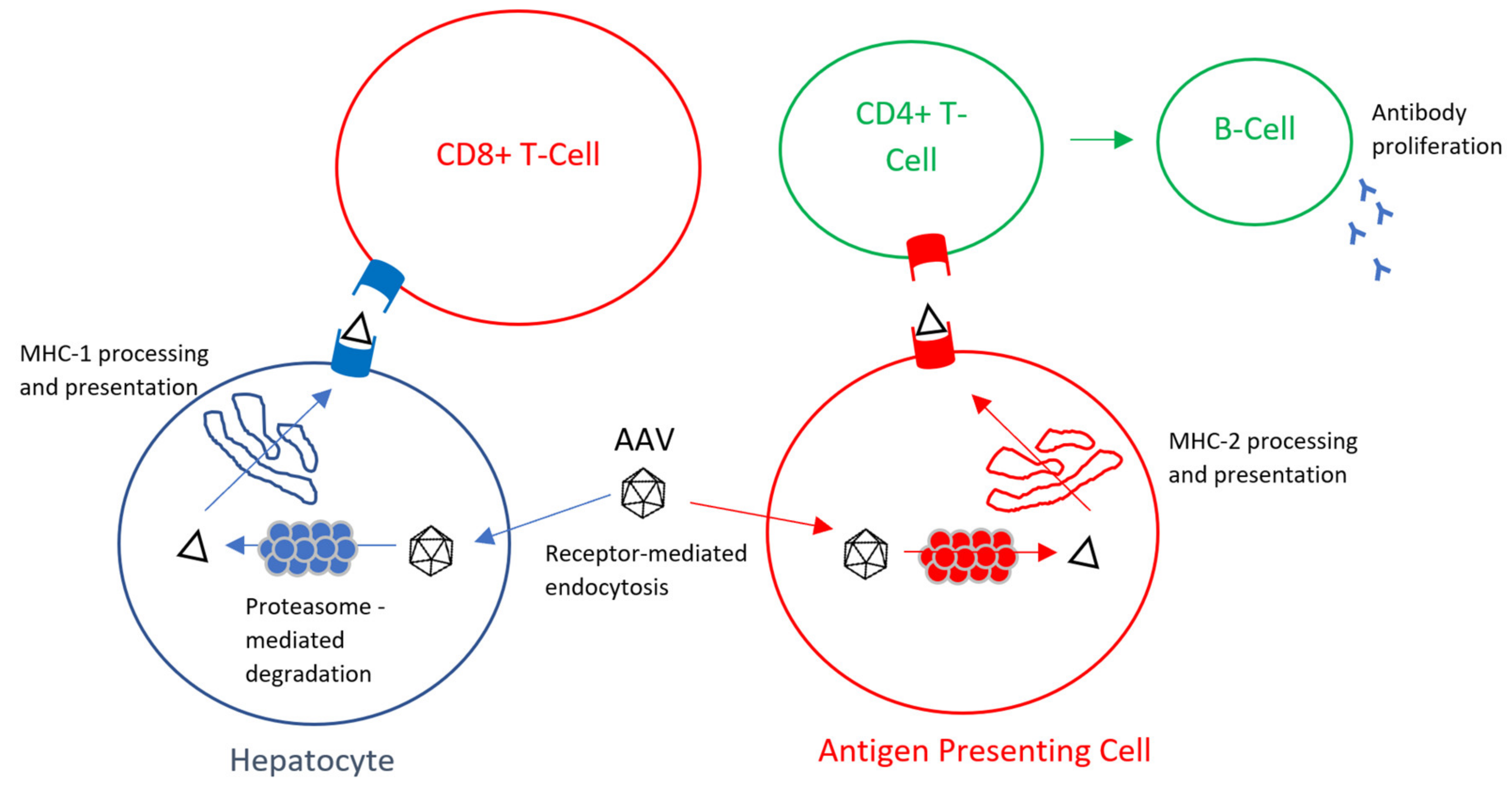{kind=link}
| MPS Type | Intervention | Company | Vector | Phase | Injection Method | Preliminary Data | Inclusion Criteria | Ref. |
|---|---|---|---|---|---|---|---|---|
| MPS I | RGX-111 | REGENXBIO Inc. | AAV9 | I/II | ICS | Expected 2nd half of 2020; inclusion criteria changed from >18 years to ≥4 months | CNS Involvement due to MPS I, 4 months and older, all sexes | [16] |
| SB-318 | Sangamo Therapeutics | AAV6/ZFN | I/II | IV | Increase in leukocyte IDUA activity into normal range. No change in plasma IDUA activity. No meaningful change in uGAG. | Clinical diagnosis of MPS I, ≥5 years, all sexes | [17] | |
| OTL-203 | Orchard Therapeutics | Autologous HSC with lentiviral vector | I/II | IV | Expected 2nd half of 2020. | Biochemically and molecularly dx MPS IH, Lansky index >80%, indication to HSCT, lack of non-heterozygous IDUA HLA-matched sibling donor, 28 days to 11 years, all sexes | [18] | |
| MPS II | RGX-121 | REGENXBIO Inc. | AAV9 | I/II | ICS | No SAEs reported. Mean reduction in CSF HS levels by 33.3% at Week 8. Stable neurocognitive development. | Documented diagnosis of MPS II AND neurocognitive testing score <77, 4 months to 5 years, male | [19] |
| SB-913 | Sangamo Therapeutics | AAV6/ZFN | I/II | IV | Small increases in IDS activity. Initial increase in plasma IDS activity, subsequent decrease due to transaminitis. No meaningful change in uGAG. | Male or female ≥5 years, clinical dx of MPS II base on clinical presentation, IDS deficiency confirmed by genetic sequencing | [20] | |
| MPS IIIA | ABO-102 | Abeona Therapeutics | AAV9 | I/II | IV | Stable or improved neurocognitive development. Sustained reduction in CSF HS. No SAEs reported. | Dx of MPS IIIA by: no detectable or reduced SGSH, genomic DNA analysis w/ mutation in SGSH, 6mo to 2 years OR >2 years w/ DQ of ≥60 | [21] |
| OTL-201 | Orchard Therapeutics | Autologous HSC with lentiviral vector | I/II | IV | None reported. | Normal cognition or mild deterioration of cognition, SGSH activity ≤10% of lower limit of normal, + normal activity of other sulfatase or mutation of SGSH, ≥3 months and ≤24 months, all sexes | [22] | |
| LYS-SAF-302 | LYSOGENE | AAVrh10 | II/III | IC | None reported. | Documented MPS IIIA diagnosis based on SGSH mutation genotyping, cognitive DQ score on BSID-III: 50% and above | [23] | |
| EGT-101 | Esteve | AAV9 | I/II | ICSF | None reported. | Under 18 years old, male and female, confirmed diagnosis of MPSIIIA | [24] | |
| MPS IIIB | ABO-101 | Abeona Therapeutics | AAV9 | III | IV | None reported. | Confirmed dx of MPSIIIB by: no detectable NAGLU in plasma, genomic DNA analysis with homo/compound heterozygous mutations in NAGLU, 6 months to 2 years OR >2 years w/ cognitive DQ ≥60 | [25] |
| MPS VI | AAV2/8.TBG.hARSB | FONDAZIONE TELETHON | AAV8 | I/II | IV | None reported. | Documented biochemical and molecular dx of MPS VI, ≥4 year, Received ERT for 12 months prior, all sexes | [26] |
| Geographic Region | Disease | Age | n | Titer Threshold | Anti-AAV Serotypes (%) | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||||||
| China (Beijing, Anhui) | N/A | <18 years | 37 | 1:10 | - | 100 | - | - | 40.5 | - | - | 67.6 | - | [48] |
| N/A | 19–30 years | 185 | 1:10 | - | 95.1 | - | - | 43.8 | - | - | 83.2 | - | ||
| N/A | 31–40 years | 162 | 1:10 | - | 96.3 | - | - | 37 | - | - | 80.9 | - | ||
| N/A | 41–56 years | 116 | 1:10 | - | 98.3 | - | - | 38.8 | - | - | 86.2 | - | ||
| United Kingdom | N/A | <6 months | 129 | 1:5 | - | - | - | - | - | - | - | 10 | - | [49] |
| N/A | 7 m–2 years | 1:5 | - | - | - | - | - | - | - | 12 | - | |||
| N/A | 3–17 years | 1:5 | - | - | - | - | - | - | - | 5 | - | |||
| N/A | >18 years | 1:5 | - | - | - | - | - | - | - | 43 | - | |||
| France | N/A | 25–64 years | 226 | Unk. | 67 | 72 | - | - | 40 | 46 | - | 38 | 47 | [50] |
| United States | MPS IIIA | 2–7 years | 16 | 1:50 | 31 | 44 | 31 | 25 | 13 | 13 | 19 | 25 | 19 | [47] |
| MPS IIIA | >8 years | 8 | 1:50 | 13 | 13 | 25 | 13 | 13 | 13 | 13 | 38 | 50 | ||
| MPS IIIB | 2–7 years | 5 | 1:50 | 40 | 20 | 40 | 20 | 20 | 20 | 20 | 20 | 20 | ||
| MPS IIIB | >8 years | 9 | 1:50 | 0 | 11 | 33 | 11 | 0 | 11 | 11 | 11 | 0 | ||
| N/A | 2–7 years | 18 | 1:50 | 6 | 17 | 22 | 22 | 6 | 17 | 11 | 17 | 6 | ||
| N/A | >8 years | 17 | 1:50 | 18 | 47 | 53 | 24 | 29 | 53 | 59 | 47 | 59 | ||
| Japan | N/A | >18 years | 85 | Unk. | 36.5 | 35.3 | - | - | 37.6 | - | - | 32.9 | 36.5 | [51] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piechnik, M.; Sawamoto, K.; Ohnishi, H.; Kawamoto, N.; Ago, Y.; Tomatsu, S. Evading the AAV Immune Response in Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 3433. https://doi.org/10.3390/ijms21103433
Piechnik M, Sawamoto K, Ohnishi H, Kawamoto N, Ago Y, Tomatsu S. Evading the AAV Immune Response in Mucopolysaccharidoses. International Journal of Molecular Sciences. 2020; 21(10):3433. https://doi.org/10.3390/ijms21103433
Chicago/Turabian StylePiechnik, Matthew, Kazuki Sawamoto, Hidenori Ohnishi, Norio Kawamoto, Yasuhiko Ago, and Shunji Tomatsu. 2020. "Evading the AAV Immune Response in Mucopolysaccharidoses" International Journal of Molecular Sciences 21, no. 10: 3433. https://doi.org/10.3390/ijms21103433
APA StylePiechnik, M., Sawamoto, K., Ohnishi, H., Kawamoto, N., Ago, Y., & Tomatsu, S. (2020). Evading the AAV Immune Response in Mucopolysaccharidoses. International Journal of Molecular Sciences, 21(10), 3433. https://doi.org/10.3390/ijms21103433