Understanding the Pathophysiology of Cerebral Amyloid Angiopathy

 ,
,  ,
,  , , ,
, , , 
{kind=link}
Abstract
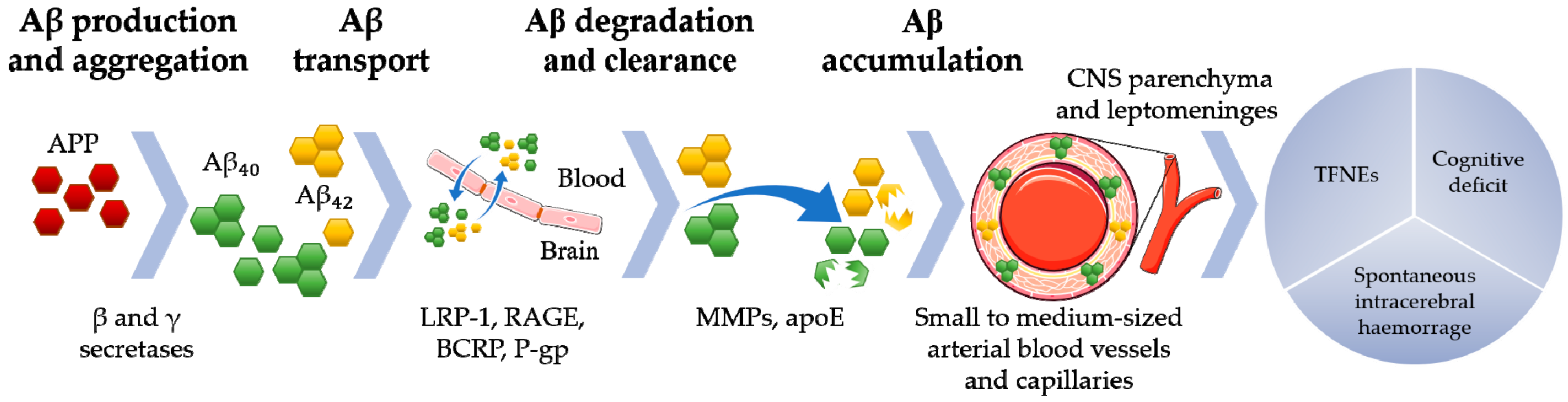
:1. Introduction
2. CAA Pathophysiological Mechanisms
2.1. Amyloid Generation and Clearance: The Role of the Neurovascular Unit
2.2. The Prion Hypothesis
2.3. Genetic Factors Involved in Aβ-CAA Formation
2.3.1. APP Genetic Variations Associated with Autosomal Dominant CAA
2.3.2. APOE Genotype in Sporadic Aβ-CAA
3. In Vitro Aβ-CAA Models
4. Animal Models
4.1. Naturally Occurring Animal Models
4.2. Transgenic Mouse Models
4.2.1. APPDutch Mice
4.2.2. APP23 Mice
4.2.3. Tg2576 Mice
4.2.4. PDAPP Mice
4.2.5. Tg-SwDI Mice
4.2.6. APP/London Mice
4.2.7. APP/PS1 Mice
4.3. Alternative Mouse Models
5. Treatment of CAA
6. Experimental Therapeutic Approaches
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAA | Cerebral amyloid angiopathy |
| CNS | Central nervous system |
| ICH | Intracerebral haemorrhage |
| Aβ | Amyloid beta protein |
| APP | Amyloid precursor protein |
| TFNEs | Transient focal neurological events |
| AD | Alzheimer’s disease |
| DW-MRI | Diffusion weighted magnetic resonance imaging |
| PET | Positron emission tomography |
| CSF | Cerebrospinal fluid |
| SMCs | Smooth muscle cells |
| BBB | Blood-brain barrier |
| LRP-1 | Low density lipoprotein receptor-related protein 1 |
| RAGE | Receptor advanced glycation end products |
| BCRP | Breast cancer resistance protein |
| P-gp | Permeability glycoprotein |
| MMPs | Matrix metallopeptidases |
| apoE | Apolipoprotein E |
| HCHWA-D | Hereditary cerebral haemorrhage with amyloidosis-Dutch type |
| NVU | Neurovascular unit |
| VSMCs | Vascular smooth muscle cells |
| CBF | Cerebral blood flow |
| ROS | Reactive oxygen species |
| HDL | High-density lipoprotein |
| BCAS | Bilateral common carotid artery stenosis |
References
- Valdés Hernández, M.D.; Qiu, X.; Wang, X.; Wiseman, S.; Sakka, E.; Maconick, L.C.; Doubal, F.; Sudlow, C.L.; Wardlaw, J.M. Interhemispheric characterization of small vessel disease imaging markers after subcortical infarct. Brain Behav. 2006, 7, e00595. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Charidimou, A. Diagnosis of cerebral amyloid angiopathy: Evolution of the Boston criteria. Stroke 2018, 49, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, S.; Romero, J.R.; Shoamanesh, A.; McKee, A.C.; Van Etten, E.; Pontes-Neto, O.; Macklin, E.A.; Ayres, A.; Auriel, E.; Himali, J.J.; et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers Dement. 2015, 11, 1480–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Xue, R.; Ge, P.; Li, M.; Wang, L.; Zheng, F.; Zhao, L.; Wang, Z.; Wang, Z.; Wang, Q.; et al. Identification of post-translational modifications of Aβ peptide in platelet membranes from patients with cerebral amyloid angiopathy. J. Neurol. Sci. 2017, 383, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Friedrich, J.O.; Greenberg, S.M.; Viswanathan, A. Core cerebrospinal fluid biomarker profile in cerebral amyloid angiopathy: A meta-analysis. Neurology 2018, 90, e754–e762. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Jaunmuktane, Z.; Baron, J.C.; Burnell, M.; Varlet, P.; Peeters, A.; Xuereb, J.; Jäger, R.; Brandner, S.; Werring, D.J. White matter perivascular spaces: An MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology 2014, 82, 57–62. [Google Scholar] [CrossRef]
- Keable, A.; Fenna, K.; Yuen, H.M.; Johnston, D.A.; Smyth, N.R.; Smith, C.; Al-Shahi Salman, R.; Samarasekera, N.; Nicoll, J.A.; Attems, J.; et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta 2016, 1862, 1037–1046. [Google Scholar] [CrossRef] [Green Version]
- Rensink, A.A.M.; de Waal, R.M.W.; Kremer, B.; Verbeek, M.M. Pathogenesis of cerebral amyloid angiopathy. Brain Res. Rev. 2003, 43, 207–223. [Google Scholar] [CrossRef]
- Smith, E.E. Cerebral amyloid angiopathy as a cause of neurodegeneration. J. Neurochem. 2018, 144, 651–658. [Google Scholar] [CrossRef]
- Kumar-Singh, S. Cerebral amyloid angiopathy: Pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 2008, 7, 67–82. [Google Scholar] [CrossRef]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, K.; Younkin, L.; Ebeling, C.; Turner, S.K.; Westaway, D.; Younkin, S.; Hsiao Ashe, K.; Carlson, G.A.; Iadecola, C. Ab1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. USA 2000, 97, 9735–9740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Olofsson, A.; Wennström, M. Amyloid-beta 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2014, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Benveniste, E.N. ELISA methodology to quantify astrocyte production of cytokines/chemokines in vitro. Methods Mol. Biol. 2012, 814, 235–249. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef]
- Hartz, A.M.; Bauer, B.; Soldner, E.L.; Wolf, A.; Boy, S.; Backhaus, R.; Mihaljevic, I.; Bogdahn, U.; Klünemann, H.H.; Schuierer, G.; et al. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 2012, 43, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Magaki, S.; Tang, Z.; Tung, S.; Williams, C.K.; Lo, D.; Yong, W.H.; Khanlou, N.; Vinters, H.V. The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol. Aging 2018, 70, 70–77. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [Green Version]
- Miners, J.S.; van Helmond, Z.; Kehoe, P.G.; Love, S. Changes with age in the activities of β-secretase and the Aβ-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 2009, 20, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, G.J.; Han, H.; Ford, A.L.; Lee, J.M. Cerebral amyloid angiopathy: Progressive disruption of the neurovascular unit. Stroke 2009, 40, S16–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldea, R.; Weller, R.O.; Wilcock, D.M.; Carare, R.O.; Richardson, G. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizari, S.; Carare, R.O.; Romero, I.A.; Hawkes, C.A. 3D reconstruction of the neurovascular unit reveals differential loss of cholinergic innervation in the cortex and hippocampus on the adult mouse brain. Front. Aging Neurosci. 2019, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Di Fede, G.; Catania, M.; Maderna, E.; Ghidoni, R.; Benussi, L.; Tonoli, E.; Giaccone, G.; Moda, F.; Paterlini, A.; Campagnani, I.; et al. Molecular subtypes of Alzheimer’s disease. Sci. Rep. 2018, 8, 3269. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef]
- Burwinkel, M.; Lutzenberger, M.; Heppner, F.L.; Schulz-Schaeffer, W.; Baier, M. Intravenous injection of beta-amyloid seeds promotes cerebral amyloid angiopathy (CAA). Acta Neuropathol. Commun. 2018, 6, 23. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, I.; Cohen, M.L.; Haik, S.; Parchi, P.; Giaccone, G.; Collins, S.J.; Kofskey, D.; Wang, H.; McLean, C.A.; Brandel, J.P.; et al. Iatrogenic Creutzfeldt-Jakob disease with amyloid-β pathology: An international study. Acta Neuropathol. Commun. 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Adams, M.E.; Jaunmuktane, Z.; Alistair Lammie, G.; Turner, B.; Wani, M.; Sawhney, I.M.S.; Houlden, H.; Mead, S.; Brandner, S.; et al. Early onset cerebral amyloid angiopathy following childhood exposure to cadaveric dura. Ann. Neurol. 2019, 85, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Quaegebeur, A.; Taipa, R.; Viana-Baptista, M.; Barbosa, R.; Koriath, C.; Sciot, R.; Mead, S.; Brandner, S. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 2018, 135, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Giaccone, G.; Maderna, E.; Marucci, G.; Catania, M.; Erbetta, A.; Chiapparini, L.; Indaco, A.; Caroppo, P.; Bersano, A.; Parati, E.; et al. Iatrogenic early onset cerebral amyloid angiopathy 30 years after cerebral trauma with neurosurgery: Vascular amyloid deposits are made up of both Aβ40 and Aβ42. Acta Neuropathol. Commun. 2019, 7, 70. [Google Scholar] [CrossRef]
- Hervé, D.; Porché, M.; Cabrejo, L.; Guidoux, C.; Tournier-Lasserve, E.; Nicolas, G.; Adle-Biassette, H.; Plu, I.; Chabriat, H.; Duyckaerts, C. Fatal Aβ cerebral amyloid angiopathy 4 decades after a dural graft at the age of 2 years. Acta Neuropathol. 2018, 135, 801–803. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Bonda, D.J.; Manjila, S.; Mehndiratta, P.; Khan, F.; Miller, B.R.; Onwuzulike, K.; Puoti, G.; Cohen, M.L.; Schonberger, L.B.; Cali, I. Human prion diseases: Surgical lessons learned from iatrogenic prion transmission. Neurosurg. Focus 2016, 41, E10. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, J.; Jucker, M.; Walker, L.C. Aβ seeds and prions: How close the fit? Prion 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Harbi, D.; Harrison, P.M. Classifying prion and prion-like phenomena. Prion 2014, 8, 161–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Frangione, B.; Rostagno, A.; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathy. Acta Neuropathol. 2009, 118, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Bugiani, O.; Giaccone, G.; Rossi, G.; Mangieri, M.; Capobianco, R.; Morbin, M.; Mazzoleni, G.; Cupidi, C.; Marcon, G.; Giovagnoli, A.; et al. Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Arch. Neurol. 2010, 67, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, D.M.A.; Davidson, Y.S.; Robinson, A.C.; Allen, N.; Hashimoto, T.; Richardson, A.; Jones, M.; Snowden, J.S.; Pendleton, N.; Potier, M.C.; et al. Patterns and severity of vascular amyloid in Alzheimer’s disease associated with duplications and missense mutations in APP gene, Down syndrome and sporadic Alzheimer’s disease. Acta Neuropathol. 2018, 136, 569–587. [Google Scholar] [CrossRef]
- Kamp, A.J.; Grand Moursel, L.; Haan, J.; Terwindt, G.M.; Lesnik Oberstein, S.A.M.J.; van Duinen, S.G.; van Roon-Mom, W.M.C. Amyloid β in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Rev. Neurosci. 2014, 25, 641–651. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Kametani, F.; Ozawa, K.; Okada, R.; Maat-Schieman, M.L.; Roos, R.A.; Iwai, K.; Mori, H. Cerebral vascular accumulation of Dutch-type Aβ42, but not wild-type Aβ42, in hereditary cerebral hemorrhage with amyloidosis, Dutch type. J. Neurosci. Res. 2007, 85, 2917–2923. [Google Scholar] [CrossRef]
- Yang, X.; Meisl, G.; Frohm, B.; Thulin, E.; Knowles, T.P.J.; Linse, S. On the role of sidechain size and charge in the aggregation of Aβ42 with familial mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E5849–E5858. [Google Scholar] [CrossRef] [Green Version]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Stürchler-Pierrat, C.; Bürki, K.; et al. Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef] [Green Version]
- Monro, O.R.; Mackic, J.B.; Yamada, S.; Segal, M.B.; Ghiso, J.; Maurer, C.; Calero, M.; Frangione, B.; Zlokovic, B.V. Substitution at codon 22 reduces clearance of Alzheimer’s amyloid-β peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol. Aging 2002, 23, 405–412. [Google Scholar] [CrossRef]
- Melchor, J.P.; McVoy, L.; van Nostrand, W.E. Change alterations of E22 enhance the pathogenic properties of the amyloid β-protein. J. Neurochem. 2000, 74, 2209–2212. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Otte-Höller, I.; David, G.; Maat-Schieman, M.L.; van den Heuvel, L.P.; Wesseling, P.; de Waal, R.M.; Verbeek, M.M. Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer’s disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol. 2001, 102, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, E.S.; Verbeek, M.M.; van der Grond, J.; Zielman, R.; van Rooden, S.; van Zwet, E.W.; van Opstal, A.M.; Haan, J.; Greenberg, S.M.; van Buchem, M.A.; et al. β-amyloid in CSF: Biomarker for preclinical amyloid angiopathy. Neurology 2017, 88, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Opstal, A.M.; van Rooden, S.; van Harten, T.; Ghariq, E.; Labadie, G.; Fotiadis, P.; Gurol, M.E.; Terwindt, G.M.; Wermer, M.J.H.; van Buchem, M.A.; et al. Cerebrovascular function in presymptomatic and symptomatic individuals with hereditary cerebral amyloid angiopathy: A case-control study. Lancet Neurol. 2017, 16, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Schouten, T.M.; de Vos, F.; van Rooden, S.; Bouts, M.J.R.J.; van Opstal, A.M.; Feis, R.A.; Terwindt, G.A.; Wermer, M.J.H.; van Buchem, M.A.; Greenberg, S.M.; et al. Multiple approaches to diffusion magnetic resonance imaging in hereditary cerebral amyloid angiopathy mutation carriers. J. Am. Heart Assoc. 2019, 8, e011288. [Google Scholar] [CrossRef]
- Schultz, A.P.; Kloet, R.W.; Sohrabi, H.R.; van der Weerd, L.; van Rooden, S.; Wermer, M.J.H.; Moursel, L.G.; Yaqub, M.; van Berckel, B.N.M.; Chatterjee, P.; et al. Amyloid imaging of Dutch-type hereditary cerabral amyloid angiopathy carriers. Ann. Neurol. 2019, 86, 616–625. [Google Scholar] [CrossRef]
- Rannikmae, K.; Samarasekera, N.; Martinez-Gonzalez, N.A.; Al-Shahi Salman, R.; Sudlow, C.L. Genetics of cerebral amyloid angiopathy: Systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 901–908. [Google Scholar] [CrossRef]
- Rannikmae, K.; Kalaria, R.N.; Greenberg, S.M.; Chui, H.C.; Schmitt, F.A.; Samarasekera, N.; Al-Shahi Salman, R.; Sudlow, C.L. APOE associations with severe CAA-associated vasculopathic changes: Collaborative meta-analysis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, S.M.; Vonsattel, J.P.; Segal, A.Z.; Chiu, R.I.; Clatworthy, A.E.; Liao, A.; Hyman, B.T.; Rebeck, G.W. Association of apolipoprotein E ε2 and vasculopathy in cerebral amyloid angiopathy. Neurology 1998, 50, 961–965. [Google Scholar] [CrossRef]
- Biffi, A.; Anderson, C.D.; Jagiella, J.M.; Schmidt, H.; Kissela, B.; Hansen, B.M.; Jimenez-Conde, J.; Pires, C.R.; Ayres, A.M.; Schwab, K.; et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: A genetic association study. Lancet Neurol. 2011, 10, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Charidimou, A.; Boulouis, G.; Greenberg, S.M.; Viswanathan, A. Cortical superficial siderosis and bleeding risk in cerebral amyloid angiopathy: A meta-analysis. Neurology 2019, 93, e2192–e2202. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Vorbrodt, A.W.; Wegiel, J. Amyloid angiopathy and blood-brain barrier changes in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1997, 826, 161–172. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J. Clin. Invest. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.; Snkhchyan, H.; Kooij, G.; van der Pol, S.; van Horssen, J.; Veerhuis, R.; Hoozemans, J.; Rozemuller, A.; de Vries, H.E. ATP-binding cassette transporters P-glycoprotein and breast cancer related protein are reduced in capillary cerebral amyloid angiopathy. Neurobiol. Aging 2014, 35, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Candela, P.; Gosselet, F.; Saint-Pol, J.; Sevin, E.; Boucau, M.C.; Boulanger, E.; Cecchelli, R.; Fenart, L. Apical-to-basolateral transport of amyloid-β peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J. Alzheimer Dis. 2010, 22, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Hartz, A.M.; Zhong, Y.; Wolf, A.; LeVine, H.; Miller, D.S.; Bauer, B. Aβ40 reduces P-glycoprotein at the blood-brain barrier through the ubiquitin-proteasome pathway. J. Neurosci. 2016, 36, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. The NADPH oxidase of endothelial cells. IUBMB Life 2000, 50, 267–269. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Zhao, X.; Wei, T. Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-beta1-42. J. Pineal. Res. 2008, 45, 157–165. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef]
- Tai, L.M.; Loughlin, A.J.; Male, D.K.; Romero, I.A. P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-beta. J. Cereb. Blood Flow Metab. 2009, 29, 1079–1083. [Google Scholar] [CrossRef]
- Yamada, K.; Hashimoto, T.; Yabuki, C.; Nagae, Y.; Tachikawa, M.; Strickland, D.K.; Liu, Q.; Bu, G.; Basak, J.M.; Holtzman, D.M.; et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J. Biol. Chem. 2008, 283, 34554–34562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazielle, N.; Ghersi-Egea, J.F.; Ghiso, J.; Dehouck, M.P.; Frangione, B.; Patlak, C.; Fenstermacher, J.; Gorevic, P. In vitro evidence that beta-amyloid peptide 1-40 diffuses across the blood-brain barrier and affects its permeability. J. Neuropathol. Exp. Neurol. 2000, 59, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A.; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.X.; He, L.Y.; Zhang, M.; Wang, F.; Liu, F.; Peng, W.X. 1,25-Dihydroxyvitamin D3 regulates expression of LRP1 and RAGE in vitro and in vivo, enhancing Aβ1-40 brain-to-blood efflux and peripheral uptake transport. Neuroscience 2016, 322, 28–38. [Google Scholar] [CrossRef]
- Sacre, S.M.; Stannard, A.K.; Owen, J.S. Apolipoprotein E (apoE) isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett. 2003, 540, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Volpato, S.; Cherubini, A.; Bandinelli, S.; Corsi, A.M.; Lauretani, F.; Guralnik, J.M.; Fellin, R.; et al. Relationship between low levels of high-density lipoprotein cholesterol and dementia in the elderly. The InChianti study. J Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Merched, A.; Xia, Y.; Visvikis, S.; Serot, J.M.; Siest, G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol. Aging. 2000, 21, 27–30. [Google Scholar] [CrossRef]
- Reitz, C.; Tang, M.X.; Schupf, N.; Manly, J.J.; Mayeux, R.; Luchsinger, J.A. Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch. Neurol. 2010, 67, 1491–1497. [Google Scholar] [CrossRef] [Green Version]
- Button, E.B.; Robert, J.; Caffrey, T.M.; Fan, J.; Zhao, W.; Wellington, C.L. HDL From an Alzheimer’s disease perspective. Curr. Opin. Lipidol. 2019, 30, 224–234. [Google Scholar] [CrossRef]
- Stukas, S.; Robert, J.; Wellington, C.L. High-density lipoproteins and cerebrovascular integrity in Alzheimer’s disease. Cell. Metab. 2014, 19, 574–591. [Google Scholar] [CrossRef] [Green Version]
- Lefterov, I.; Fitz, N.F.; Cronican, A.A.; Fogg, A.; Lefterov, P.; Kodali, R.; Wetzel, R.; Koldamova, R. Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J. Biol. Chem. 2010, 285, 36945–36957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Button, E.B.; Boyce, G.K.; Wilkinson, A.; Stukas, S.; Hayat, A.; Fan, J.; Wadsworth, B.J.; Robert, J.; Martens, K.M.; Wellington, C.L. ApoA-I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid-associated astrocyte reactivity in APP/PS1 mice. Alzheimers Res. Ther. 2019, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, T.L.; Cao, D.; Lu, H.; Mans, R.A.; Su, Y.R.; Jungbauer, L.; Linton, M.R.F.; Fazio, S.; LaDu, M.J.; Li, L. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J. Biol. Chem. 2010, 285, 36958–36968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, J.; Stukas, S.; Button, E.; Cheng, W.H.; Lee, M.; Fan, J.; Wilkinson, A.; Kulic, I.; Wright, S.D.; Wellington, C.L. Reconstituted high-density lipoproteins acutely reduce soluble brain Aβ levels in symptomatic APP/PS1 mice. Biochim. Biophys. Acta 2016, 1862, 1027–1036. [Google Scholar] [CrossRef]
- Fernández-de Retana, S.; Montañola, A.; Marazuela, P.; De La Cuesta, M.; Batlle, A.; Fatar, M.; Grudzenski, S.; Montaner, J.; Hernández-Guillamon, M. Intravenous treatment with human recombinant ApoA-I Milano reduces beta amyloid cerebral deposition in the APP23-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 60, 116–128. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of β-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. eLife 2017, 6, e29595. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Stukas, S.; Boyce, G.K.; Gibbs, E.; Cowan, C.M.; Gilmour, M.; Cheng, W.H.; Soo, S.K.; Yuen, B.; et al. High-density lipoproteins suppress Aβ-induced PBMC adhesion to human endothelial cells in bioengineered vessels and in monoculture. Mol. Neurodegener. 2017, 12, 60. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Martin, E.M.; McAlary, L.; Gidden, Z.; Gilmour, M.; Boyce, G.; Caffrey, T.M.; Agbay, A.; Clark, A.; et al. Cerebrovascular amyloid angiopathy in bioengineered vessels is reduced by high-density lipoprotein particles enriched in apolipoprotein E. Mol. Neurodegener. 2020, 15, 23. [Google Scholar] [CrossRef]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Morinaga, A.; Hasegawa, K.; Nomura, R.; Ookoshi, T.; Ozawa, D.; Goto, Y.; Yamada, M.; Naiki, H. Critical role of interfaces and agitation on the nucleation of Aβ amyloid fibrils at low concentrations of Aβ monomers. Biochim. Biophys. Acta 2010, 1804, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Ozawa, D.; Ookoshi, T.; Naiki, H. Surface-bound basement membrane components accelerate amyloid-β peptide nucleation in air-free wells: An in vitro model of cerebral amyloid angiopathy. Biochim. Biophys. Acta 2013, 1834, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Hasegawa, K.; Nomura, R.; Arishima, H.; Kikuta, K.I.; Yamashita, T.; Inoue, Y.; Ueda, M.; Ando, Y.; Wilson, M.R.; et al. Apolipoprotein E and clusterin inhibit the early phase of amyloid-β aggregation in an in vitro model of cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.M.; Ma, J.F. The role of amyloid β clearance in cerebral amyloid angiopathy: More potential therapeutic targets. Transl. Neurodegener. 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainsworth, A.H.; Allan, S.M.; Boltze, J.; Cunningham, C.; Farris, C.; Head, E.; Ihara, M.; Isaacs, J.D.; Kalaria, R.N.; Lesnik Oberstein, S.A.; et al. Translational models for vascular cognitive impairment: A review including larger species. BMC Med. 2017, 15, 16. [Google Scholar] [CrossRef]
- Herzig, M.C.; van Nostrand, W.E.; Jucker, M. Mechanism of cerebral beta-amyloid angiopathy: Murine and cellular models. Brain Pathol. 2006, 16, 40–54. [Google Scholar] [CrossRef]
- Jäkel, L.; van Nostrand, W.E.; Nicoll, J.A.R.; Werring, D.J.; Verbeek, M.M. Animal models of cerebral amyloid angiopathy. Clin. Sci. 2017, 131, 2469–2488. [Google Scholar] [CrossRef]
- Schmidt, F.; Boltze, J.; Jäger, C.; Hofmann, S.; Willems, N.; Seeger, J.; Härtig, W.; Stolzing, A. Detection and quantification of β-amyloid, pyroglutamyl Aβ, and Tau in aged canines. J. Neuropathol. Exp. Neurol. 2015, 74, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Schelle, J.; Wegenast-Braun, B.M.; Fritschi, S.K.; Kaeser, S.A.; Jährling, N.; Eicke, D.; Skodras, A.; Beschorner, N.; Obermueller, U.; Häsler, L.M.; et al. Early Aβ reduction prevents progression of cerebral amyloid angiopathy. Ann. Neurol. 2019, 86, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.H.; Abramowski, D.; Sturchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. USA 1999, 96, 14088–14093. [Google Scholar] [CrossRef] [Green Version]
- Winkler, D.T.; Bondolfi, L.; Herzig, M.C.; Jann, L.; Calhoun, M.E.; Wiederhold, K.H.; Tolnay, M.; Staufenbiel, M.; Jucker, M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 2001, 21, 1619–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.; Grigoryan, M.M.; Kilday, K.; Sumbria, R.K.; Vasilevko, V.; van Ryn, J.; Cribbs, D.H.; Paganini-Hill, A.; Fisher, M.J. Effects of dabigatran in mouse models of aging and cerebral amyloid angiopathy. Front Neurol. 2019, 10, 966. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid β-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Xu, F.; Davis, J.; Otte-Höller, I.; Verbeek, M.M.; van Nostrand, W.E. Cerebral microvascular amyloid β protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid β precursor protein. Am. J. Pathol. 2005, 167, 505–515. [Google Scholar] [CrossRef]
- Van Dorpe, J.; Smeijers, L.; Dewachter, I.; Nuyens, D.; Spittaels, K.; Van Den Haute, C.; Mercken, M.; Moechars, D.; Laenen, I.; Kuiperi, C.; et al. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am. J. Pathol. 2000, 157, 1283–1298. [Google Scholar] [CrossRef]
- Van Groen, T.; Kiliaan, A.J.; Kadish, I. Deposition of mouse amyloid beta in human APP/PS1 double and single AD model transgenic mice. Neurobiol. Dis. 2006, 23, 653–662. [Google Scholar] [CrossRef]
- Jiao, S.S.; Bu, X.L.; Liu, Y.H.; Zhu, C.; Wang, Q.H.; Shen, L.L.; Liu, C.H.; Wang, Y.R.; Yao, X.Q.; Wang, Y.J. Sex dimorphism profile of Alzheimer’s disease-type pathologies in an APP/PS1 mouse model. Neurotox. Res. 2016, 29, 256–266. [Google Scholar] [CrossRef]
- Van Veluw, S.J.; Frosch, M.P.; Scherlek, A.A.; Lee, D.; Greenberg, S.M.; Bacskai, B.J. In vivo characterization of spontaneous microhemorrhage formation in mice with cerebral amyloid angiopathy. J. Cereb. Blood Flow Metab. 2020. [Google Scholar] [CrossRef]
- Mazza, M.; Marano, G.; Traversi, G.; Bria, P.; Mazza, S. Primary cerebral blood flow deficiency and Alzheimer’s disease: Shadows and lights. J. Alzheimers Dis. 2011, 23, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Yamamoto, T.; Kalaria, R.N.; Senzaki, H.; Maki, T.; Hase, Y.; Kitamura, A.; Washida, K.; Yamada, M.; Ito, H.; et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 2012, 123, 381–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, Y.; Yamashita, T.; Nakano, Y.; Sun, Z.; Shang, J.; Feng, T.; Morihara, R.; Fukui, Y.; Ohta, Y.; Hishikawa, N.; et al. Chronic cerebral hypoperfusion accelerates Alzheimer’s disease pathology with cerebrovascular remodeling in a novel mouse model. J. Alzheimers Dis. 2016, 53, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.T.; Poulet, R.; Di Pardo, A.; Cifelli, G.; Maffei, A.; Vecchione, C.; Passarelli, F.; Landolfi, A.; Carullo, P.; Lembo, G. β-amyloid deposition in brain is enhanced in mouse models of arterial hypertension. Neurobiol. Aging 2009, 30, 222–228. [Google Scholar] [CrossRef]
- Cifuentes, D.; Poittevin, M.; Dere, E.; Broquères-You, D.; Bonnin, P.; Benessiano, J.; Pocard, M.; Mariani, J.; Kubis, N.; Merkulova-Rainon, T.; et al. Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension 2015, 65, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Sudduth, T.L.; Weekman, E.M.; Brothers, H.M.; Braun, K.; Wilcock, D.M. β-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimers Res. Ther. 2014, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Hemphill, J.C., 3rd; Greenberg, S.M.; Anderson, C.S.; Becker, K.; Bendok, B.R.; Cushman, M.; Fung, G.L.; Goldstein, J.N.; Macdonald, R.L.; Mitchell, P.H.; et al. Guidelines for the management of spontaneous intracerebral hemorrhage: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2015, 46, 2032–2060. [Google Scholar] [CrossRef] [Green Version]
- Arima, H.; Tzourio, C.; Anderson, C.; Woodward, M.; Bousser, M.G.; MacMahon, S.; Neal, B.; Chalmers, J.; PROGRESS Collaborative Group. Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: The PROGRESS trial. Stroke 2010, 41, 394–396. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Anderson, C.D.; Battey, T.W.; Ayres, A.M.; Greenberg, S.M.; Viswanathan, A.; Rosand, J. Association between blood pressure control and risk of recurrent intracerebral hemorrhage. JAMA 2015, 314, 904–912. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Shams, S.; Calvet, D.; Shoamanesh, A.; International META-MICROBLEEDS Initiative. Meta-analysis methodology in the microbleeds field: The relevance of the clinical question and study quality in choosing the most appropriate model. J. Neurol. Sci. 2017, 381, 348–349. [Google Scholar] [CrossRef]
- Weber, S.A.; Patel, R.K.; Lutsep, H.L. Cerebral amyloid angiopathy: Diagnosis and potential therapies. Expert. Rev. Neurother. 2018, 18, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.R.; O’Neill, S.M.; Pozdnyakov, N.; Pan, F.; Caouette, D.; Pi, Y.; Wood, K.M.; Volfson, D.; Cirrito, J.R.; Han, B.H.; et al. Passive immunotherapy targeting amyloid-β reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain 2016, 139, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Leurent, C.; Goodman, J.A.; Zhang, Y.; He, P.; Polimeni, J.R.; Gurol, M.E.; Lindsay, M.; Frattura, L.; Sohur, U.S.; Viswanathan, A.; et al. Ponezumab trial study group, Greenberg SM2. Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann. Clin. Transl. Neurol. 2019, 6, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Okamoto, Y.; Carare, R.O.; Hase, Y.; Hattori, Y.; Hawkes, C.A.; Saito, S.; Yamamoto, Y.; Terasaki, Y.; Ishibashi-Ueda, H.; et al. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular β-amyloid. Ann. Clin. Transl. Neurol. 2014, 1, 519–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Hamazaki, T.S.; Sugaya, M.; Fukuda, S.; Chan, T.; Tamura-Nakano, M.; Sato, S.; Okochi, H. Cilostazol improves lymphatic function by inducing proliferation and stabilization of lymphatic endothelial cells. J. Dermatol. Sci. 2014, 74, 150–158. [Google Scholar] [CrossRef]
- Sumbria, R.K.; Vasilevko, V.; Grigoryan, M.M.; Paganini-Hill, A.; Kim, R.; Cribbs, D.H.; Fisher, M.J. Effects of phosphodiesterase 3A modulation on murine cerebral microhemorrhages. J. Neuroinflamm. 2017, 14, 114. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Yamamoto, Y.; Maki, T.; Hattori, Y.; Ito, H.; Mizuno, K.; Harada-Shiba, M.; Kalaria, R.N.; Fukushima, M.; Takahashi, R.; et al. Taxifolin inhibits amyloid-β oligomer formation and fully restores vascular integrity and memory in cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2017, 5, 26. [Google Scholar] [CrossRef]
- Inoue, T.; Yamakage, H.; Tanaka, M.; Kusakabe, T.; Shimatsu, A.; Satoh-Asahara, N. Oxytocin suppresses inflammatory responses induced by lipopolysaccharide through inhibition of the eIF-2α–ATF4 pathway in mouse microglia. Cells 2019, 8, 527. [Google Scholar] [CrossRef] [Green Version]
- Iwata, N.; Mizukami, H.; Shirotani, K.; Takaki, Y.; Muramatsu, S.; Lu, B.; Gerard, N.P.; Gerard, C.; Ozawa, K.; Saido, T.C. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-β peptide in mouse brain. J. Neurosci. 2004, 24, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polis, B.; Gurevich, V.; Assa, M.; Samson, A.O. Norvaline restores the BBB integrity in a mouse model of Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, M.; Pankiewicz, J.; Scholtzova, H.; Ripellino, J.A.; Li, Y.; Schmidt, S.D.; Mathews, P.M.; Fryer, J.D.; Holtzman, D.M.; Sigurdsson, E.M.; et al. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am. J. Pathol. 2004, 165, 937–948. [Google Scholar] [CrossRef]
- Yang, J.; Ji, Y.; Mehta, P.; Bates, K.A.; Sun, Y.; Wisniewski, T. Blocking the apolipoprotein E/amyloid-β interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J. Alzheimers Dis. 2011, 24, 269–285. [Google Scholar] [CrossRef]
- Liu, S.; Park, S.; Allington, G.; Prelli, F.; Sun, Y.; Martá-Ariza, M.; Scholtzova, H.; Biswas, G.; Brown, B.; Verghese, P.B.; et al. Targeting apolipoprotein E/Amyloid β binding by peptoid CPO_Aβ17-21 P ameliorates Alzheimer’s disease related pathology and cognitive decline. Sci. Rep. 2017, 7, 8009. [Google Scholar] [CrossRef] [Green Version]
- Mahley, R.W.; Huang, Y. Apolipoprotein E sets the stage: Response to injury triggers neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef] [Green Version]
- Brodbeck, J.; McGuire, J.; Liu, Z.; Meyer-Franke, A.; Balestra, M.E.; Jeong, D.E.; Pleiss, M.; McComas, C.; Hess, F.; Witter, D.; et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 2011, 286, 17217–17226. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, T.; Drummond, E. Future horizons in Alzheimer’s disease research. Prog. Mol. Biol. Transl. Sci. 2019, 168, 223–241. [Google Scholar] [CrossRef]
- Walker, D.G.; Dalsing-Hernandez, J.E.; Lue, L.F. Human postmortem brain-derived cerebrovascular smooth muscle cells express all genes of the classical complement pathway: A potential mechanism for vascular damage in cerebral amyloid angiopathy and Alzheimer’s disease. Microvasc. Res. 2008, 75, 411–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, S.A.; Katusic, Z.S. Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. J. Cereb. Blood Flow Metab. 2020, 40, 392–403. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Perkins, A.; Cisternas, P.; Muñoz, B.; Taylor, X.; You, Y.; Garringer, H.J.; Oblak, A.L.; Atwood, B.K.; Vidal, R.; et al. Tau as a mediator of neurotoxicity associated to cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2019, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersano, A.; Scelzo, E.; Pantoni, L.; Morotti, A.; Erbetta, A.; Chiapparini, L.; Vitali, P.; Giaccone, G.; Caroppo, P.; Catania, M.; et al. Discovering the Italian phenotype of cerebral amyloid angiopathy (CAA): The SENECA Project. Neurol. Sci. 2020. online ahead of print. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatti, L.; Tinelli, F.; Scelzo, E.; Arioli, F.; Di Fede, G.; Obici, L.; Pantoni, L.; Giaccone, G.; Caroppo, P.; Parati, E.A.; et al. Understanding the Pathophysiology of Cerebral Amyloid Angiopathy. Int. J. Mol. Sci. 2020, 21, 3435. https://doi.org/10.3390/ijms21103435
Gatti L, Tinelli F, Scelzo E, Arioli F, Di Fede G, Obici L, Pantoni L, Giaccone G, Caroppo P, Parati EA, et al. Understanding the Pathophysiology of Cerebral Amyloid Angiopathy. International Journal of Molecular Sciences. 2020; 21(10):3435. https://doi.org/10.3390/ijms21103435
Chicago/Turabian StyleGatti, Laura, Francesca Tinelli, Emma Scelzo, Francesco Arioli, Giuseppe Di Fede, Laura Obici, Leonardo Pantoni, Giorgio Giaccone, Paola Caroppo, Eugenio Agostino Parati, and et al. 2020. "Understanding the Pathophysiology of Cerebral Amyloid Angiopathy" International Journal of Molecular Sciences 21, no. 10: 3435. https://doi.org/10.3390/ijms21103435
APA StyleGatti, L., Tinelli, F., Scelzo, E., Arioli, F., Di Fede, G., Obici, L., Pantoni, L., Giaccone, G., Caroppo, P., Parati, E. A., & Bersano, A. (2020). Understanding the Pathophysiology of Cerebral Amyloid Angiopathy. International Journal of Molecular Sciences, 21(10), 3435. https://doi.org/10.3390/ijms21103435