Harmful Iron-Calcium Relationship in Pantothenate kinase Associated Neurodegeneration

 , ,
, ,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
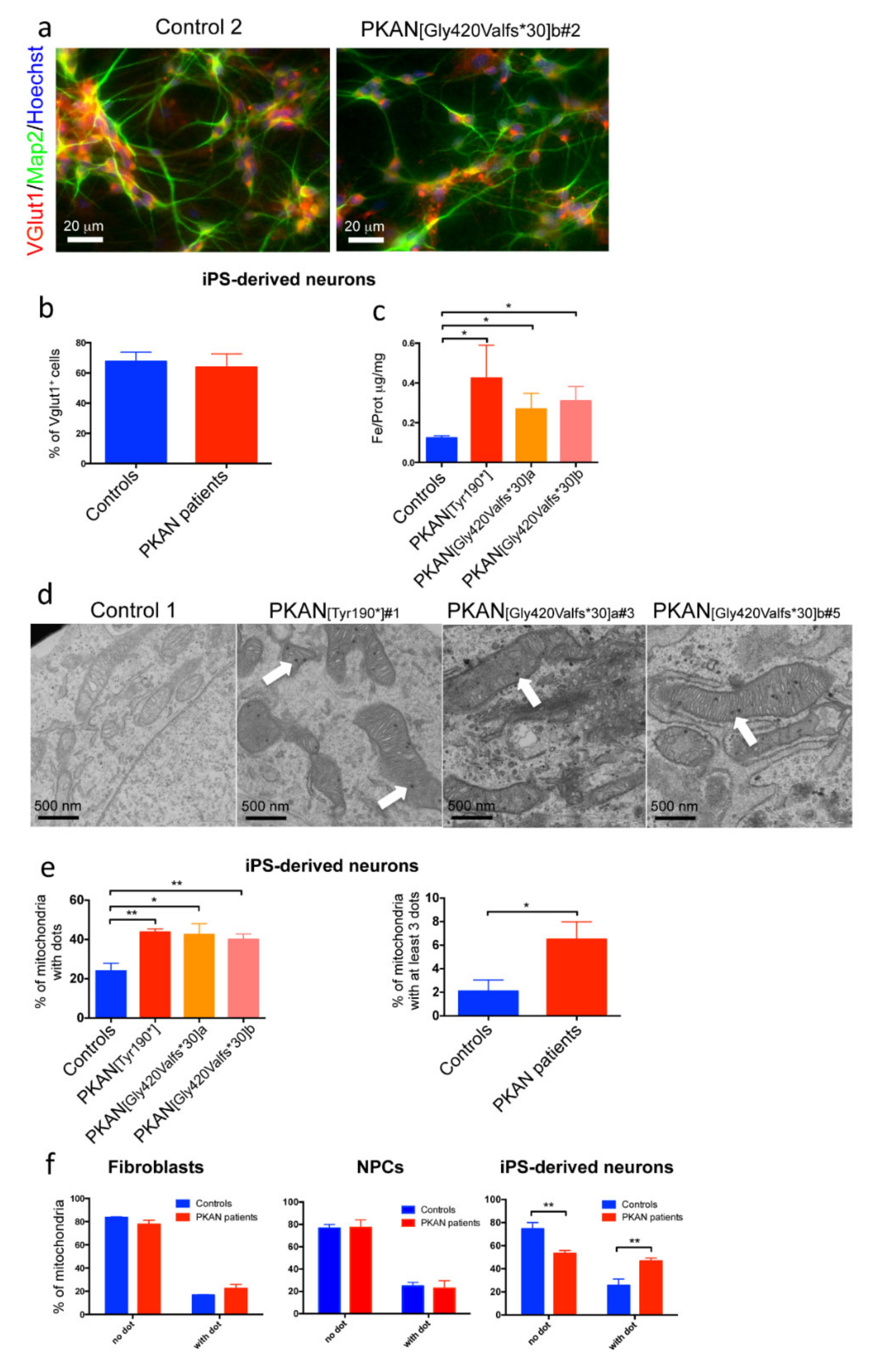
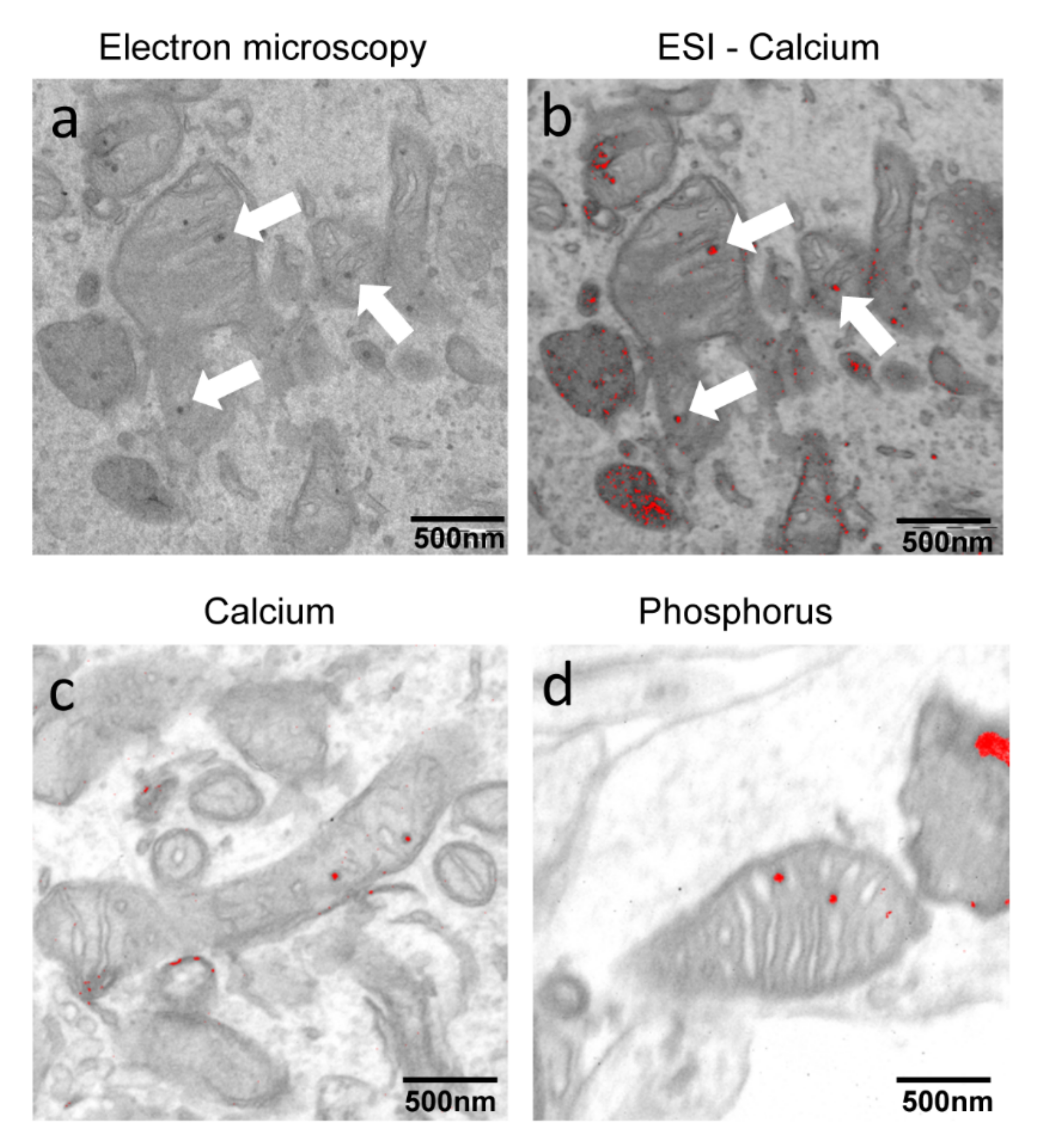
2.1. PKAN iPS-Derived Neurons Show Altered Iron Content and Deposit of Calcium Phosphate in Mitochondria
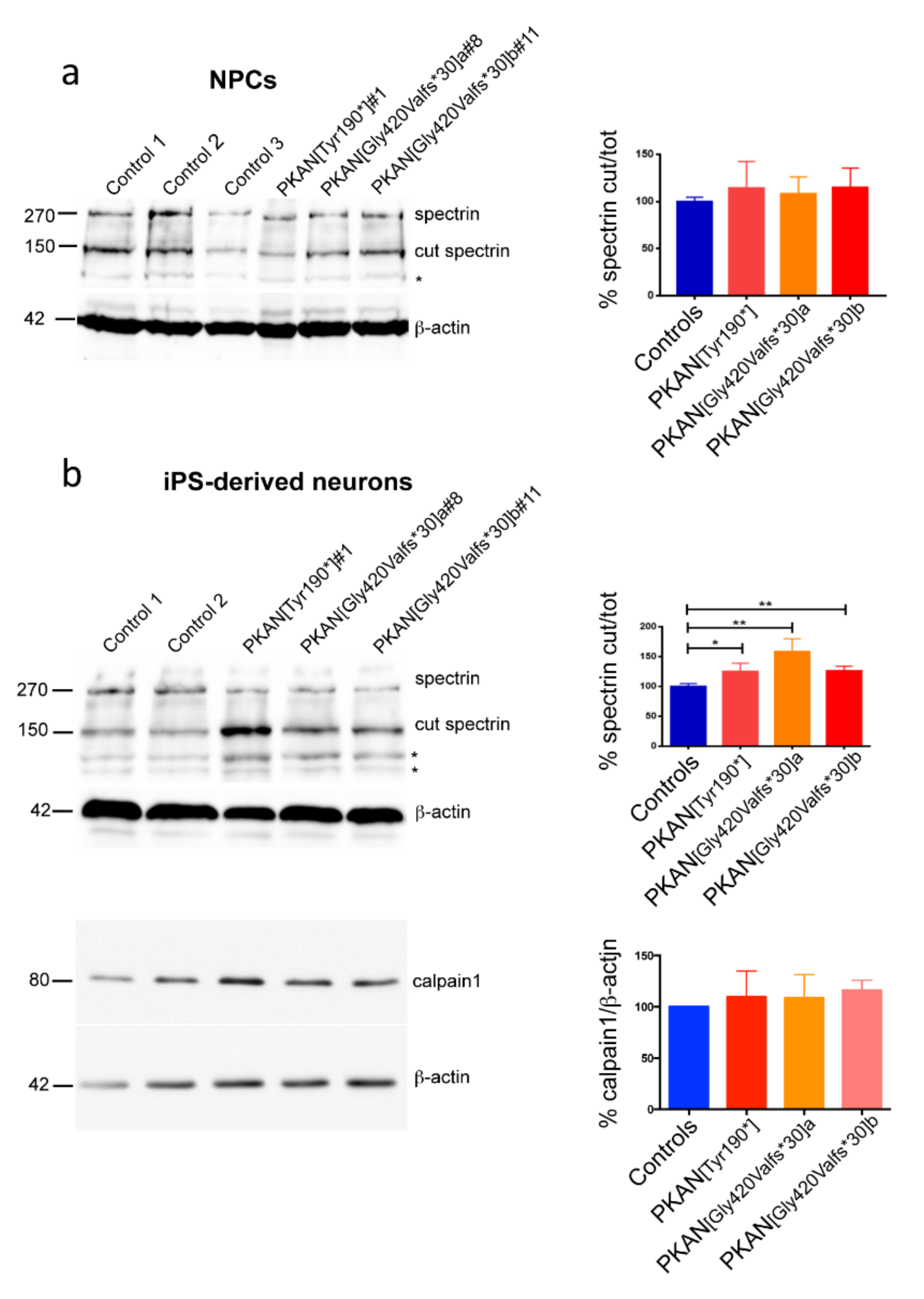
2.2. Calpain1 Activity Increase in PKAN iPS-Derived Neurons
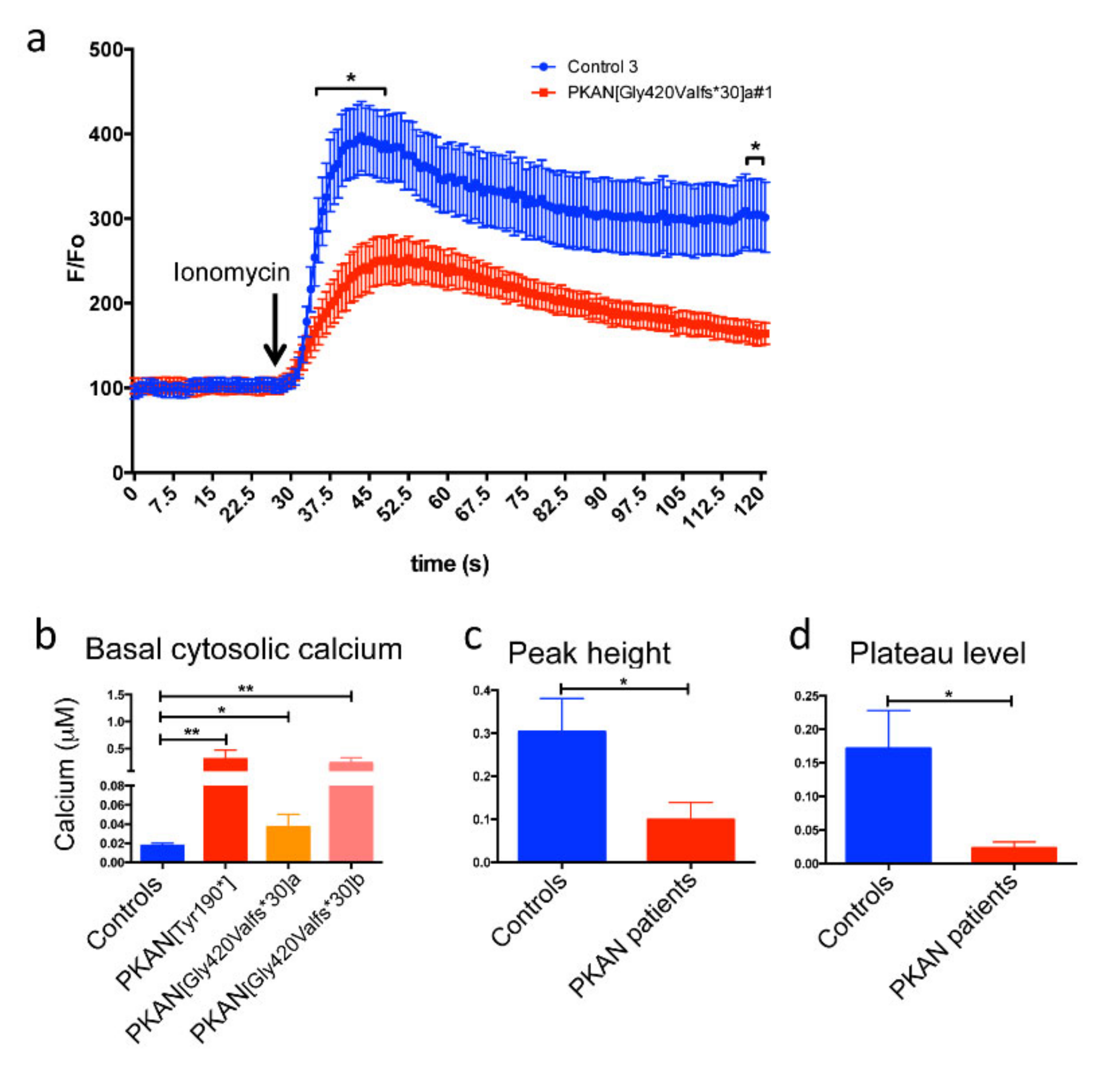
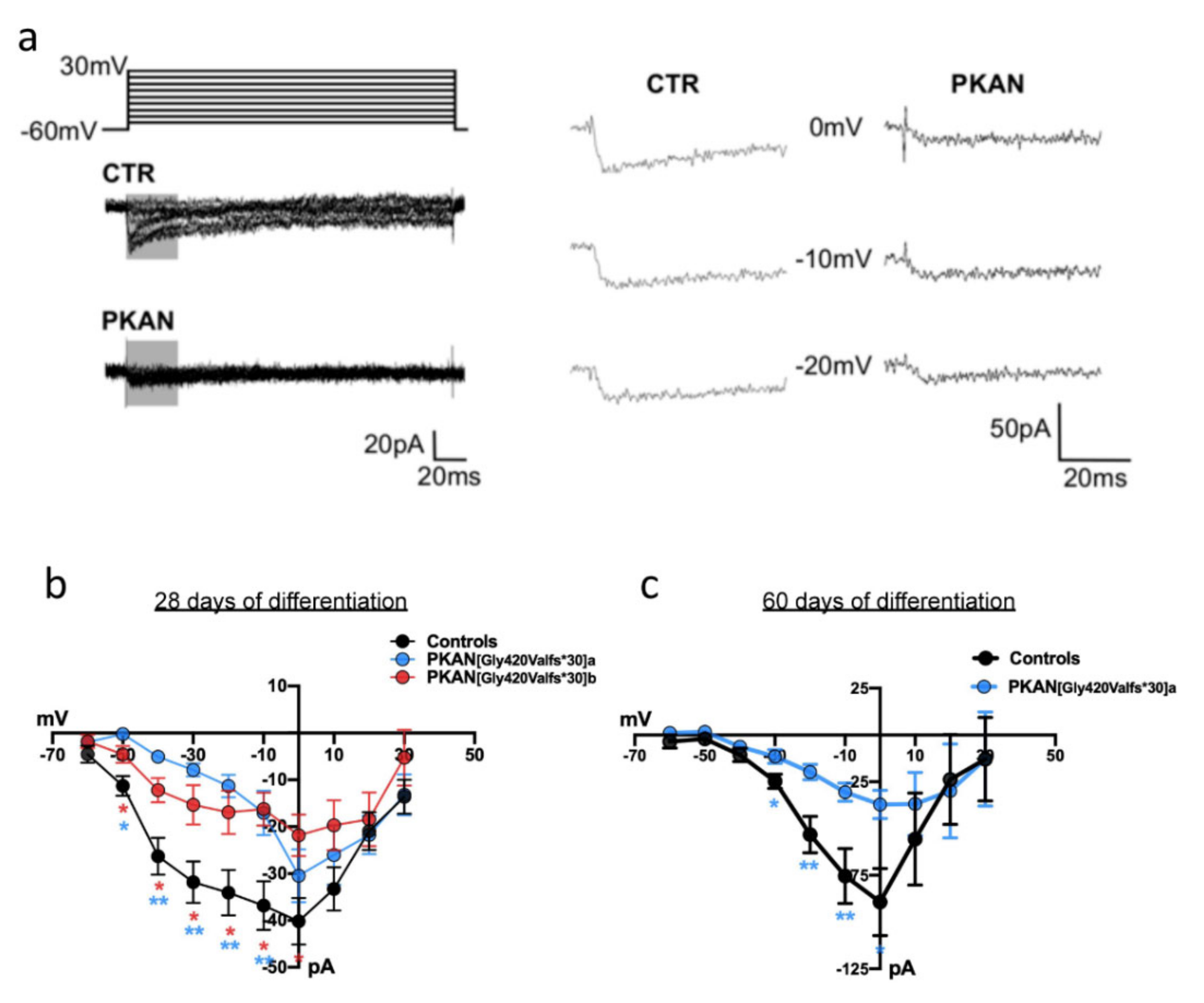
2.3. The Cytosol of PKAN iPS-Derived Neurons Shows Less Calcium Influx than Controls
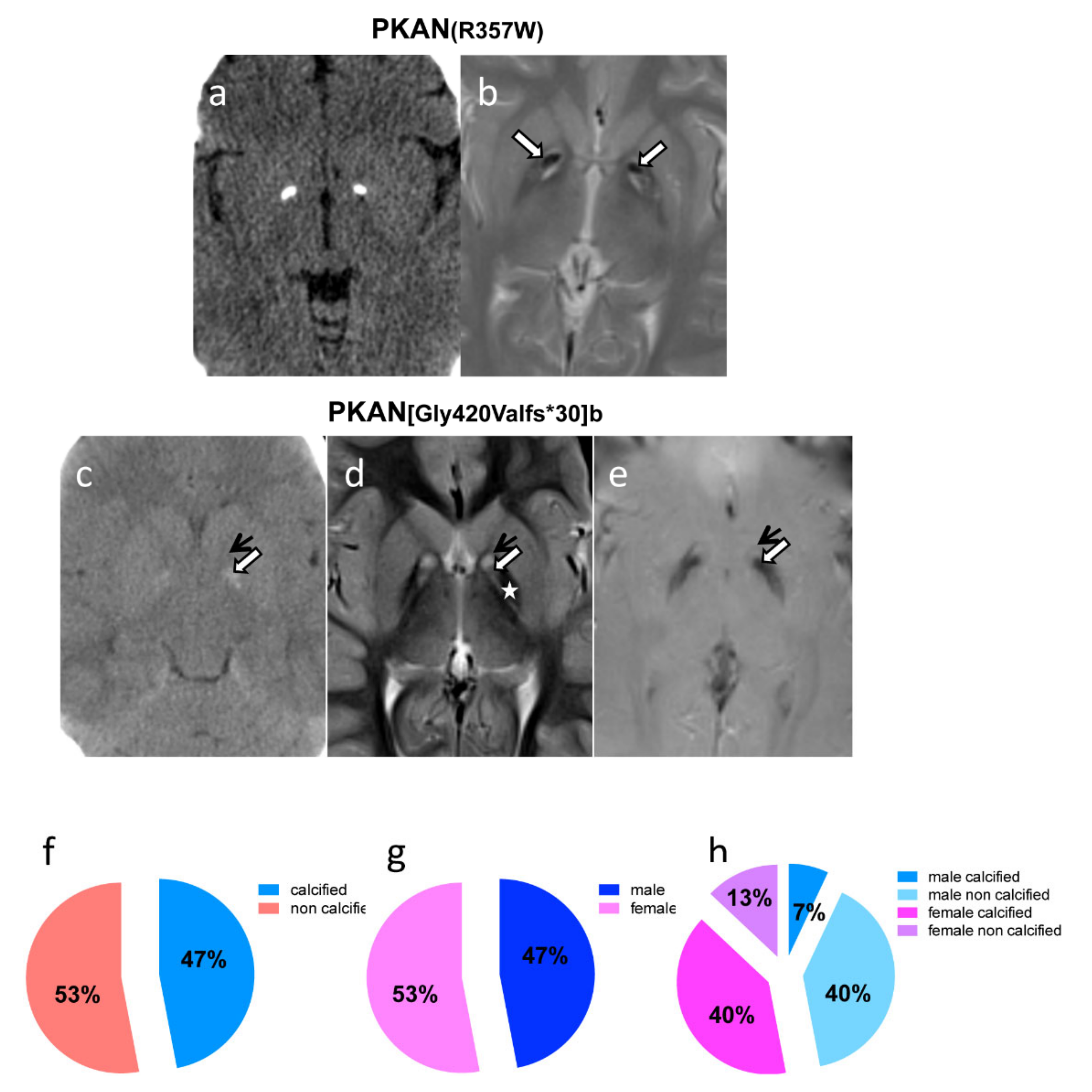
2.4. Neuroimaging Confirms Calcium Accumulation In Vivo
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Fibroblasts Culture and iPSC Generation
4.3. Generation of Human Neuronal Precursors Cells and iPS-Derived Neurons
4.4. Immunofluorescence
4.5. Immunoblotting
4.6. Electron Microscopy (EM) and Electron Spectroscopic Imaging (ESI)
4.7. Ca2+ Imaging
4.8. Electrophysiology
4.9. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
4.10. Brain MRI and CT Scan
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CoA | Coenzyme A |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| iPSC | Induced Pluripotent Stem Cells |
| NBIA | Neurodegeneration with Brain Iron Accumulation |
| PANK2 | pantothenate kinase 2 |
| PKAN | Pantothenate Kinase-Associated Neurodegeneration |
References
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with Brain Iron Accumulation. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 293–305. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Westaway, S.K.; Levinson, B.; Zhou, B.; Johnson, M.A.; Ching, K.H.L.; Gitschier, J. Genetic, Clinical, and Radiographic Delineation of Hallervorden-Spatz Syndrome. N. Engl. J. Med. 2003, 348, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Hogarth, P.; Kurian, M.A.; Gregory, A.; Csányi, B.; Zagustin, T.; Kmiec, T.; Wood, P.; Klucken, A.; Scalise, N.; Sofia, F.; et al. Consensus Clinical Management Guideline for Pantothenate Kinase-Associated Neurodegeneration (PKAN). Mol. Genet. Metab. 2017, 120, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.A. Neurodegeneration with Brain Iron Accumulation. Curr. Neurol. Neurosci. Rep. 2016, 16, 9. [Google Scholar] [CrossRef]
- Hartig, M.B.; Hörtnagel, K.; Garavaglia, B.; Zorzi, G.; Kmiec, T.; Klopstock, T.; Rostasy, K.; Svetel, M.; Kostic, V.S.; Schuelke, M.; et al. Genotypic and Phenotypic Spectrum of PANK2 Mutations in Patients with Neurodegeneration with Brain Iron Accumulation. Ann. Neurol. 2006, 59, 248–256. [Google Scholar] [CrossRef]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A Novel Pantothenate Kinase Gene (PANK2) Is Defective in Hallervorden-Spatz Syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; D’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate Kinase-Associated Neurodegeneration: Altered Mitochondria Membrane Potential and Defective Respiration in Pank2 Knock-out Mouse Model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef] [Green Version]
- Kotzbauer, P.T.; Truax, A.C.; Trojanowski, J.Q.; Lee, V.M.Y. Altered Neuronal Mitochondrial Coenzyme A Synthesis in Neurodegeneration with Brain Iron Accumulation Caused by Abnormal Processing, Stability, and Catalytic Activity of Mutant Pantothenate Kinase 2. J. Neurosci. 2005, 25, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Pecchio, A.; Garcia, M.; Leonardi, R.; Jackowski, S. Compartmentalization of Mammalian Pantothenate Kinases. PLoS ONE 2012, 7, e49509. [Google Scholar] [CrossRef] [Green Version]
- Klopstock, T.; Tricta, F.; Neumayr, L.; Karin, I.; Zorzi, G.; Fradette, C.; Kmieć, T.; Büchner, B.; Steele, H.E.; Horvath, R.; et al. Safety and Efficacy of Deferiprone for Pantothenate Kinase-Associated Neurodegeneration: A Randomised, Double-Blind, Controlled Trial and an Open-Label Extension Study. Lancet Neurol. 2019, 18, 631–642. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine Treatment Is Effective in Recovering the Disease Phenotype Induced by Ketogenic Diet in a Pantothenate Kinase-Associated Neurodegeneration Mouse Model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; Van Der Want, J.J.; Hayflick, S.; Reijngoud, D.J.; Kayser, O.; Sibon, O.C.M. Pantethine Rescues a Drosophila Model for Pantothenate Kinase-Associated Neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, B.; Baratashvili, M.; Van Der Zwaag, M.; Kanon, B.; Colombelli, C.; Lambrechts, R.A.; Schaap, O.; Nollen, E.A.; Podgoršek, A.; Kosec, G.; et al. Extracellular 4′-Phosphopantetheine Is a Source for Intracellular Coenzyme A Synthesis. Nat. Chem. Biol. 2015, 11, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Hogarth, P.; Placzek, A.; Gregory, A.M.; Fox, R.; Zhen, D.; Hamada, J.; van der Zwaag, M.; Lambrechts, R.; Jin, H.; et al. 4′-Phosphopantetheine Corrects CoA, Iron, and Dopamine Metabolic Defects in Mammalian Models of PKAN. EMBO Mol. Med. 2019, 11, e10489. [Google Scholar] [CrossRef] [PubMed]
- Christou, Y.P.; Tanteles, G.A.; Kkolou, E.; Ormiston, A.; Konstantopoulos, K.; Beconi, M.; Marshall, R.D.; Plotkin, H.; Kleopa, K.A. Open-Label Fosmetpantotenate, a Phosphopantothenate Replacement Therapy in a Single Patient with Atypical PKAN. Case Rep. Neurol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Finazzi, D. Neurodegeneration with Brain Iron Accumulation: Update on Pathogenic Mechanisms. Front. Pharmacol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A Corrects Pathological Defects in Human Neurons of PANK 2-associated Neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef]
- Klopstock, T.; Escolar, M.L.; Marshall, R.D.; Perez-Dueñas, B.; Tuller, S.; Videnovic, A.; Greblikas, F. The FOsmetpantotenate Replacement Therapy (FORT) Randomized, Double-Blind, Placebo-Controlled Pivotal Trial: Study Design and Development Methodology of a Novel Primary Efficacy Outcome in Patients with Pantothenate Kinase-Associated Neurodegeneration. Clin. Trials 2019, 16, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Budde, T.; Meuth, S.; Pape, H.C. Calcium-Dependent Inactivation of Neuronal Calcium Channels. Nat. Rev. Neurosci. 2002, 3, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Zavala, G.; Castillo, K.; Aguirre, P.; Hidalgo, C.; Núñez, M.T. Effect of Iron on the Activation of the MAPK/ERK Pathway in PC12 Neuroblastoma Cells. Biol. Res. 2006, 39, 189–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, P.; Humeres, A.; Elgueta, C.; Kirkwood, A.; Hidalgo, C.; Núñez, M.T. Iron Mediates N-Methyl-D-Aspartate Receptor-Dependent Stimulation of Calcium-Induced Pathways and Hippocampal Synaptic Plasticity. J. Biol. Chem. 2011, 286, 13382–13392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, C.; Carrasco, M.A.; Muñoz, P.; Núñez, M.T. A Role for Reactive Oxygen/Nitrogen Species and Iron on Neuronal Synaptic Plasticity. Antioxid. Redox Signal. 2007, 9, 245–255. [Google Scholar] [CrossRef]
- Hidalgo, C.; Núñez, M.T. Calcium, Iron and Neuronal Function. IUBMB Life 2007, 59, 280–285. [Google Scholar] [CrossRef]
- SanMartín, C.D.; Paula-Lima, A.C.; García, A.; Barattini, P.; Hartel, S.; Núñez, M.T.; Hidalgo, C. Ryanodine Receptor-Mediated Ca2+ Release Underlies Iron-Induced Mitochondrial Fission and Stimulates Mitochondrial Ca2+ Uptake in Primary Hippocampal Neurons. Front. Mol. Neurosci. 2014, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Zheng, F.; Ye, X.; Li, X.; Zhao, Q.; Lin, Z.; Hu, Y.; Wang, J. Basal Ganglia Calcification and Novel Compound Heterozygous Mutations in the PANK2 Gene in a Chinese Boy with Classic Pantothenate Kinase-Associated Neurodegeneration: A Case Report. Medicine (Baltimore) 2018, 97, e0316. [Google Scholar] [CrossRef]
- Zheng, J.; Ramirez, V.D. Purification and Identification of an Estrogen Binding Protein from Rat Brain: Oligomycin Sensitivity-Conferring Protein (OSCP), a Subunit of Mitochondrial FOF1-ATP Synthase/ATPase. J. Steroid Biochem. Mol. Biol. 1999, 68, 65–75. [Google Scholar] [CrossRef]
- Vega-Vela, N.E.; Osorio, D.; Avila-Rodriguez, M.; Gonzalez, J.; García-Segura, L.M.; Echeverria, V.; Barreto, G.E. L-Type Calcium Channels Modulation by Estradiol. Mol. Neurobiol. 2017, 54, 4996–5007. [Google Scholar] [CrossRef]
- Núñez, M.T.; Hidalgo, C. Noxious Iron–Calcium Connections in Neurodegeneration. Front. Neurosci. 2019, 13, 48. [Google Scholar] [CrossRef] [Green Version]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium Signaling in Neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef]
- Westenberger, A.; Balck, A.; Klein, C. Primary Familial Brain Calcifications: Genetic and Clinical Update. Curr. Opin. Neurol. 2019, 32, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial Iron and Energetic Dysfunction Distinguish Fibroblasts and Induced Neurons from Pantothenate Kinase-Associated Neurodegeneration Patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traoré, M.; Habarou, F.; Bole-Feysot, C.; Nitschké, P.; Ottolenghi, C.; et al. Impaired Transferrin Receptor Palmitoylation and Recycling in Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indrigo, M.; Papale, A.; Orellana, D.; Brambilla, R. Lentiviral Vectors to Study the Differential Function of ERK1 and ERK2 MAP Kinases. Methods Mol. Biol. 2010, 661, 205–220. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient’s Code (Sex) | PANK2 Mutations $ (cDNA) | PANK2 Mutations $ (Protein) | Date of CT Scan | Globi Pallida Calcifications | DBS | Date of Brain MRI | Age of Disease Onset |
|---|---|---|---|---|---|---|---|
| mt4245 (f) | c.[821_822delCT]; [1561G > A] | p.[Leu275Valfs*16]; [Gly521Arg] | 2014 | bilateral rock calcifications | DBS | 2006; 2013 | 2 years |
| HA44 (f) | c.[1069C > T]; [1069C > T] | p.[Arg35Trp]; [Arg35Trp] | 2013; 2015 | bilateral rock calcifications | DBS | 2008; 2009 | 24 years |
| HA143 (f) | c.[965A > G]; [1561G > A] | p.[Glu322Gly]; [Gly521Arg] | 2015 | bilateral rock calcifications | DBS | 2006; 2015 | 16 years |
| HA31 (f) | c.[821_822delCT]; [del ex 1-4] | p.[Leu275Valfs*16]; [not translated] | 2000 | bilateral rock calcifications | 2001; 2002 | 1 year | |
| HA26 (m) | c.[1441C > T]; c.[966G > T] | p.[Arg481*]; p.[Glu322Asp] | 2017 | no calcifications | 2014; 2015; 2017 | 17 years | |
| HA35 (m) | c.[790C > T]; [790C > T] | p.[Arg264Trp]; [Arg264Trp] | 2015 | bilateral calcifications | NA | 2008; 2009 | NA |
| HA101 (f) | c.[1259delG]; [1259delG] | p.[Gly420Valfs*30]; [Gly420Valfs*30] | 2007 | bilateral calcifications | 2007; 2008; 2009 | 4 years | |
| HA102 (m) | c.[1259delG]; [1259delG] | p.[Gly420Valfs*30]; [Gly420Valfs*30] | 2007 | no calcifications | 2007; 2008; 2009; 2013 | 7 years | |
| HA134 (m) | c.[1499A > T]; [621-1G > A] | p.[Asn500Ile]; ? | 2008 | no calcifications | 2004; 2008 | 2 years | |
| mt4597 (m) | c.[683T > C]; [683T > C] | p.[Phe228Ser]; [Phe228Ser] | 2013 | no calcifications | DBS | 2006; 2008; 2011 | 2 years |
| HA167 (m) | c.[821_822delCT]; [965A > G] | p.[Leu275Valfs*16]; [Glu322Gly] | 2011 | no calcifications | 2009; 2010; 2011 | 9 years | |
| HA316 (f) | c.[821_822delCT]; [1441C > T] | p.[Leu275Valfs*16]; [Arg481*] | 2015 | bilateral slight calcifications | 2005 (normal); 2007 (pathological iron); 2013 | 18 months | |
| HA185 (f) | c.[36T > A]; [137A > T] | p.[His12Gln]; [Asp46Val] | 2010 | no calcifications | 2010; 2011; 2012; 2015; 2019 | 12 years | |
| BDM862 (f) | c.[1236-1G > A]; [1561G > A] | p.?;[Gly521Arg] | 2015; 2017 | no calcifications | 2015; 2016 | 5 years | |
| LDM709 (m) | c.[982-1G > A]; [1561G > A] | p.?;[Gly521Arg] | 2019 | no calcifications | 2017; 2019 | 8 years |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santambrogio, P.; Ripamonti, M.; Paolizzi, C.; Panteghini, C.; Carecchio, M.; Chiapparini, L.; Raimondi, M.; Rubio, A.; Di Meo, I.; Cozzi, A.; et al. Harmful Iron-Calcium Relationship in Pantothenate kinase Associated Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3664. https://doi.org/10.3390/ijms21103664
Santambrogio P, Ripamonti M, Paolizzi C, Panteghini C, Carecchio M, Chiapparini L, Raimondi M, Rubio A, Di Meo I, Cozzi A, et al. Harmful Iron-Calcium Relationship in Pantothenate kinase Associated Neurodegeneration. International Journal of Molecular Sciences. 2020; 21(10):3664. https://doi.org/10.3390/ijms21103664
Chicago/Turabian StyleSantambrogio, Paolo, Maddalena Ripamonti, Chiara Paolizzi, Celeste Panteghini, Miryam Carecchio, Luisa Chiapparini, Marzia Raimondi, Alicia Rubio, Ivano Di Meo, Anna Cozzi, and et al. 2020. "Harmful Iron-Calcium Relationship in Pantothenate kinase Associated Neurodegeneration" International Journal of Molecular Sciences 21, no. 10: 3664. https://doi.org/10.3390/ijms21103664
APA StyleSantambrogio, P., Ripamonti, M., Paolizzi, C., Panteghini, C., Carecchio, M., Chiapparini, L., Raimondi, M., Rubio, A., Di Meo, I., Cozzi, A., Taverna, S., De Palma, G., Tiranti, V., & Levi, S. (2020). Harmful Iron-Calcium Relationship in Pantothenate kinase Associated Neurodegeneration. International Journal of Molecular Sciences, 21(10), 3664. https://doi.org/10.3390/ijms21103664