Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. AD-Associated Epilepsy and Epileptic Seizures
3. GSK-3 Is Involved in the Development of Seizures in AD
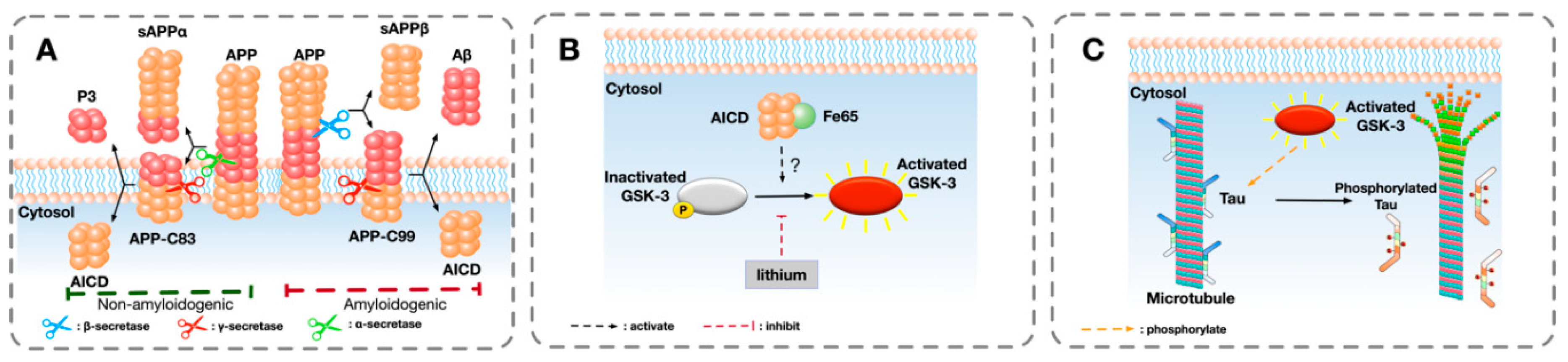
4. Putative Interaction Between APP and GSK-3
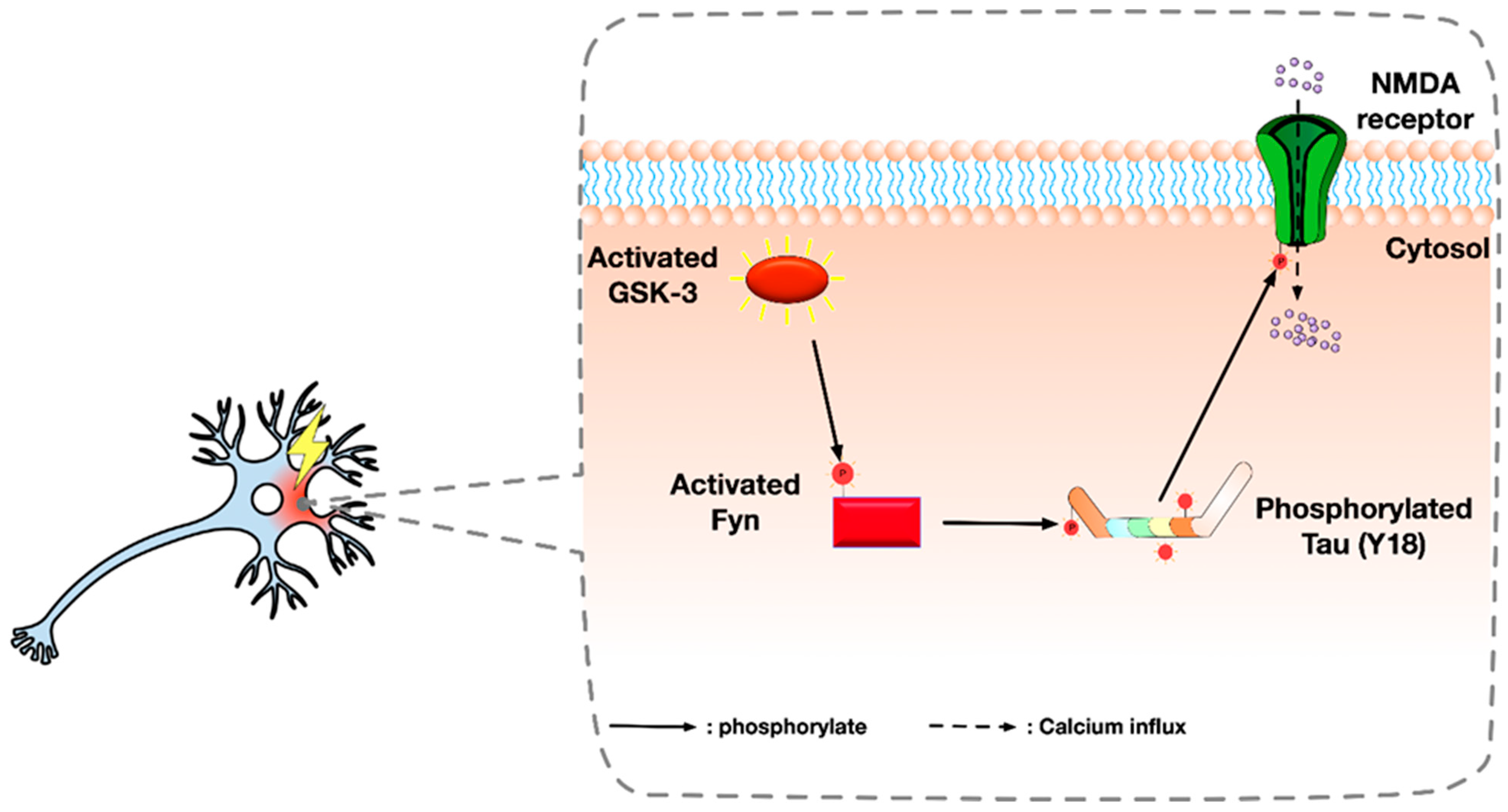
5. Alternative Roles of GSK-3 in AD and Seizures
6. Future Research Directions
Author Contributions
Funding
Conflicts of Interest
References
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 2007, 3, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Hesdorffer, D.C.; Hauser, W.A.; Annegers, J.F.; Kokmen, E.; Rocca, W.A. Dementia and adult-onset unprovoked seizures. Neurology 1996, 46, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.A.; Morris, M.L.; Heston, L.L.; Anderson, V.E. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986, 36, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, M.F.; Morris, J.C.; Ashkin, K.; Coben, L.A. Advanced Alzheimer’s disease is a risk factor for late-onset seizures. Arch Neurol. 1990, 47, 847–850. [Google Scholar] [CrossRef]
- Bakker, A.; Krauss, G.L.; Albert, M.S.; Speck, C.L.; Jones, L.R.; Stark, C.E.; Yassa, M.A.; Bassett, S.S.; Shelton, A.L.; Gallagher, M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 2012, 74, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Rabinowicz, A.L.; Starkstein, S.E.; Leiguarda, R.C.; Coleman, A.E. Transient epileptic amnesia in dementia: A treatable unrecognized cause of episodic amnestic wandering. Alzheimer Dis. Assoc. Disord. 2000, 14, 231–233. [Google Scholar] [CrossRef]
- Scharfman, H.E. Alzheimer’s disease and epilepsy: Insight from animal models. Future Neurol 2012, 7, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.A. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009, 47, 289–299. [Google Scholar]
- Sperfeld, A.D.; Collatz, M.B.; Baier, H.; Palmbach, M.; Storch, A.; Schwarz, J.; Tatsch, K.; Reske, S.; Joosse, M.; Heutink, P.; et al. FTDP-17: An early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann. Neurol. 1999, 46, 708–715. [Google Scholar] [CrossRef]
- Garcia-Cabrero, A.M.; Guerrero-Lopez, R.; Giraldez, B.G.; Llorens-Martin, M.; Avila, J.; Serratosa, J.M.; Sanchez, M.P. Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis. 2013, 58, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.; Verret, L.; Juan, C.; Remaud, J.; Halley, H.; Rampon, C.; Dahan, L. Early onset of hypersynchronous network activity and expression of a marker of chronic seizures in the Tg2576 mouse model of Alzheimer’s disease. PLoS ONE 2015, 10, e0119910. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fulop, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkanen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef]
- Dunning, C.J.; McGauran, G.; Willen, K.; Gouras, G.K.; O’Connell, D.J.; Linse, S. Direct High Affinity Interaction between Abeta42 and GSK3alpha Stimulates Hyperphosphorylation of Tau. A New Molecular Link in Alzheimer’s Disease? ACS Chem. Neurosci. 2016, 7, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Muyllaert, D.; Kremer, A.; Jaworski, T.; Borghgraef, P.; Devijver, H.; Croes, S.; Dewachter, I.; Van Leuven, F. Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav. 2008, 7 (Suppl. S1), 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.C.; Dove, G.; Cascino, G.D.; Petersen, R.C. Recurrent seizures in patients with dementia: Frequency, seizure types, and treatment outcome. Epilepsy Behav. 2009, 14, 118–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, W.A.; Annegers, J.F.; Kurland, L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993, 34, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Cabrejo, L.; Guyant-Marechal, L.; Laquerriere, A.; Vercelletto, M.; De la Fourniere, F.; Thomas-Anterion, C.; Verny, C.; Letournel, F.; Pasquier, F.; Vital, A.; et al. Phenotype associated with APP duplication in five families. Brain 2006, 129, 2966–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.E.; Bird, T.D. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- de Jonghe-Rouleau, A.P.; Pot, A.M.; de Jonghe, J.F. Self-injurious behaviour in nursing home residents with dementia. Int. J. Geriatr. Psychiatry 2005, 20, 651–657. [Google Scholar] [CrossRef]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef]
- Vossel, K.A.; Ranasinghe, K.G.; Beagle, A.J.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; Van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Ziyatdinova, S.; Ronnback, A.; Gurevicius, K.; Miszczuk, D.; Graff, C.; Winblad, B.; Pitkanen, A.; Tanila, H. Increased Epileptiform EEG Activity and Decreased Seizure Threshold in Arctic APP Transgenic Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Corbett, B.F.; Leiser, S.C.; Ling, H.P.; Nagy, R.; Breysse, N.; Zhang, X.; Hazra, A.; Brown, J.T.; Randall, A.D.; Wood, A.; et al. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 7020–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Born, H.A.; Kim, J.Y.; Savjani, R.R.; Das, P.; Dabaghian, Y.A.; Guo, Q.; Yoo, J.W.; Schuler, D.R.; Cirrito, J.R.; Zheng, H.; et al. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer’s disease. J. Neurosci. 2014, 34, 3826–3840. [Google Scholar] [CrossRef] [PubMed]
- Vogt, D.L.; Thomas, D.; Galvan, V.; Bredesen, D.E.; Lamb, B.T.; Pimplikar, S.W. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol. Aging 2011, 32, 1725–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan, V.; Gorostiza, O.F.; Banwait, S.; Ataie, M.; Logvinova, A.V.; Sitaraman, S.; Carlson, E.; Sagi, S.A.; Chevallier, N.; Jin, K.; et al. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl. Acad. Sci. USA 2006, 103, 7130–7135. [Google Scholar] [CrossRef] [Green Version]
- Saganich, M.J.; Schroeder, B.E.; Galvan, V.; Bredesen, D.E.; Koo, E.H.; Heinemann, S.F. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J. Neurosci. 2006, 26, 13428–13436. [Google Scholar] [CrossRef]
- Volicer, L.; Smith, S.; Volicer, B.J. Effect of seizures on progression of dementia of the Alzheimer type. Dementia 1995, 6, 258–263. [Google Scholar] [CrossRef]
- Lott, I.T.; Doran, E.; Nguyen, V.Q.; Tournay, A.; Movsesyan, N.; Gillen, D.L. Down syndrome and dementia: Seizures and cognitive decline. J. Alzheimers Dis. 2012, 29, 177–185. [Google Scholar] [CrossRef]
- Nygaard, H.B.; Wagner, A.F.; Bowen, G.S.; Good, S.P.; MacAvoy, M.G.; Strittmatter, K.A.; Kaufman, A.C.; Rosenberg, B.J.; Sekine-Konno, T.; Varma, P.; et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Holmes, G.L.; Lenck-Santini, P.P. Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav. 2006, 8, 504–515. [Google Scholar] [CrossRef]
- Kleen, J.K.; Scott, R.C.; Holmes, G.L.; Lenck-Santini, P.P. Hippocampal interictal spikes disrupt cognition in rats. Ann. Neurol. 2010, 67, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, H.B.; Kaufman, A.C.; Sekine-Konno, T.; Huh, L.L.; Going, H.; Feldman, S.J.; Kostylev, M.A.; Strittmatter, S.M. Brivaracetam, but not ethosuximide, reverses memory impairments in an Alzheimer’s disease mouse model. Alzheimers Res. Ther. 2015, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.Y.; Zheng, C.Y.; Zou, M.M.; Zhu, J.W.; Zhang, Y.; Wang, J.; Liu, C.F.; Li, Q.F.; Xiao, Z.C.; Li, S.; et al. Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol. Aging 2014, 35, 2713–2725. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Wang, B.R.; Tian, Y.Y.; Xu, J.; Gao, L.; Zhao, S.L.; Jiang, T.; Xie, H.G.; Zhang, Y.D. Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci. Ther. 2013, 19, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, K.; Shiratsuchi, A.; Sato, S.; Omori, A.; Arioka, M.; Kobayashi, S.; Uchida, T.; Imahori, K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993, 325, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Tian, F.F.; Chen, J.M.; Guo, T.H.; Ma, Y.F.; Fang, J.; Dang, J.; Song, M.Y. GSK-3beta may be involved in hippocampal mossy fiber sprouting in the pentylenetetrazole-kindling model. Mol. Med. Rep. 2013, 8, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Ryder, J.; Su, Y.; Liu, F.; Li, B.; Zhou, Y.; Ni, B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem. Biophys. Res. Commun. 2003, 312, 922–929. [Google Scholar] [CrossRef]
- Avila, J.; Wandosell, F.; Hernandez, F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 2010, 10, 703–710. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9 (Suppl. S3), 309–317. [Google Scholar] [CrossRef]
- Ishiguro, K.; Takamatsu, M.; Tomizawa, K.; Omori, A.; Takahashi, M.; Arioka, M.; Uchida, T.; Imahori, K. Tau protein kinase I converts normal tau protein into A68-like component of paired helical filaments. J. Biol. Chem. 1992, 267, 10897–10901. [Google Scholar]
- Wagner, U.; Utton, M.; Gallo, J.M.; Miller, C.C. Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. J. Cell Sci. 1996, 109, 1537–1543. [Google Scholar]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [Green Version]
- Chong, F.P.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Tau Proteins and Tauopathies in Alzheimer’s Disease. Cell Mol. Neurobiol. 2018, 38, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Mucke, L. Tau Phosphorylation-Much More than a Biomarker. Neuron 2016, 92, 265–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hall, A.M.; Kelinske, M.; Roberson, E.D. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol. Aging 2014, 35, 2617–2624. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Shen, Y.; Shultz, S.R.; Nguyen, A.; Hovens, C.; Adlard, P.A.; Bush, A.I.; Chan, J.; Kwan, P.; O’Brien, T.J.; et al. Accelerated kindling epileptogenesis in Tg4510 tau transgenic mice, but not in tau knockout mice. Epilepsia 2017, 58, e136–e141. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Zheng, P.; Wright, D.K.; Dezsi, G.; Braine, E.; Nguyen, T.; Corcoran, N.M.; Johnston, L.A.; Hovens, C.M.; Mayo, J.N.; et al. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 2016, 139, 1919–1938. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.C.; Nguyen, T.; Corcoran, N.M.; Velakoulis, D.; Chen, T.; Grundy, R.; O’Brien, T.J.; Hovens, C.M. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol. Dis. 2012, 45, 897–901. [Google Scholar] [CrossRef]
- Bennecib, M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 2000, 485, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Menezes, M.E.; Pannell, L.K.; Mulekar, M.S.; Honkanen, R.E.; Shevde, L.A.; Samant, R.S. DNAJB6 chaperones PP2A mediated dephosphorylation of GSK3beta to downregulate beta-catenin transcription target, osteopontin. Oncogene 2012, 31, 4472–4483. [Google Scholar] [CrossRef] [Green Version]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [Green Version]
- Saraswati, A.P.; Ali Hussaini, S.M.; Krishna, N.H.; Babu, B.N.; Kamal, A. Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions. Eur. J. Med. Chem. 2018, 144, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Martinez, A. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2014–2015). Expert Opin. Ther. Pat. 2017, 27, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Lesort, M.; Jope, R.S.; Johnson, G.V. Insulin transiently increases tau phosphorylation: Involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 1999, 72, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Shen, F.; Tian, K.; Wang, M.; Xi, Y.; Li, J.; Huang, Z. Triptolide induces oxidative damage in NRK-52E cells through facilitating Nrf2 degradation by ubiquitination via the GSK-3beta/Fyn pathway. Toxicol In Vitro 2019, 58, 187–194. [Google Scholar] [CrossRef]
- Kojima, N.; Ishibashi, H.; Obata, K.; Kandel, E.R. Higher seizure susceptibility and enhanced tyrosine phosphorylation of N-methyl-D-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem. 1998, 5, 429–445. [Google Scholar]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef]
- Miyamoto, T.; Stein, L.; Thomas, R.; Djukic, B.; Taneja, P.; Knox, J.; Vossel, K.; Mucke, L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 2017, 12, 41. [Google Scholar] [CrossRef]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Torrance, M.; Adame, A.; Mante, M.; Bar-on, P.; Rose, J.B.; Crews, L.; Masliah, E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 2007, 27, 1981–1991. [Google Scholar] [CrossRef]
- Ishizawa, T.; Sahara, N.; Ishiguro, K.; Kersh, J.; McGowan, E.; Lewis, J.; Hutton, M.; Dickson, D.W.; Yen, S.H. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am. J. Pathol. 2003, 163, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kim, E.M.; Lee, J.P.; Park, C.H.; Kim, S.; Seo, J.H.; Chang, K.A.; Yu, E.; Jeong, S.J.; Chong, Y.H.; et al. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J. 2003, 17, 1951–1953. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.A.; Kim, H.S.; Ha, T.Y.; Ha, J.W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.P.; Park, C.H.; Kim, S.; Baik, T.K.; et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, K.A.; Pimplikar, S.W. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J. Cell Biol. 2005, 171, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Pimplikar, S.W. Aging and excitotoxic stress exacerbate neural circuit reorganization in amyloid precursor protein intracellular domain transgenic mice. Neurobiol. Aging 2011, 32, 2320 e1-9. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef] [Green Version]
- von Rotz, R.C.; Kohli, B.M.; Bosset, J.; Meier, M.; Suzuki, T.; Nitsch, R.M.; Konietzko, U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 2004, 117, 4435–4448. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, D.M.; Miller, C.C. The FE65 proteins and Alzheimer’s disease. J. Neurosci. Res. 2008, 86, 744–754. [Google Scholar] [CrossRef]
- Jaworski, T.; Dewachter, I.; Lechat, B.; Gees, M.; Kremer, A.; Demedts, D.; Borghgraef, P.; Devijver, H.; Kugler, S.; Patel, S.; et al. GSK-3alpha/beta kinases and amyloid production in vivo. Nature 2011, 480, E4–E5. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Takahashi, M.; Hayashi, S.; Kakita, A.; Wakabayashi, K.; Fukuda, M.; Kameyama, S.; Tanaka, R.; Takahashi, H.; Nawa, H. Patients with temporal lobe epilepsy show an increase in brain-derived neurotrophic factor protein and its correlation with neuropeptide Y. Brain Res 1999, 818, 579–582. [Google Scholar] [CrossRef]
- Gangarossa, G.; Sakkaki, S.; Lory, P.; Valjent, E. Mouse hippocampal phosphorylation footprint induced by generalized seizures: Focus on ERK, mTORC1 and Akt/GSK-3 pathways. Neuroscience 2015, 311, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Biel, N.; Canudas, A.M.; Camins, A.; Pallas, M. Kainate induces AKT, ERK and cdk5/GSK3beta pathway deregulation, phosphorylates tau protein in mouse hippocampus. Neurochem. Int. 2007, 50, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.P.; Santorufo, G.; Brilli, E.; Borrelli, E.; Bozzi, Y. Kainic acid-induced seizures activate GSK-3beta in the hippocampus of D2R-/- mice. Neuroreport 2010, 21, 846–850. [Google Scholar] [CrossRef]
- Engel, T.; Gomez-Sintes, R.; Alves, M.; Jimenez-Mateos, E.M.; Fernandez-Nogales, M.; Sanz-Rodriguez, A.; Morgan, J.; Beamer, E.; Rodriguez-Matellan, A.; Dunleavy, M.; et al. Bi-directional genetic modulation of GSK-3beta exacerbates hippocampal neuropathology in experimental status epilepticus. Cell Death Dis. 2018, 9, 969. [Google Scholar] [CrossRef]
- Joyce, J.N.; Myers, A.J.; Gurevich, E. Dopamine D2 receptor bands in normal human temporal cortex are absent in Alzheimer’s disease. Brain Res. 1998, 784, 7–17. [Google Scholar] [CrossRef]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.A.; Halatsch, M.E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; et al. Phosphoinositide 3-kinases upregulate system xc(-) via eukaryotic initiation factor 2alpha and activating transcription factor 4—A pathway active in glioblastomas and epilepsy. Antioxid Redox Signal 2014, 20, 2907–2922. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.N.; Kaeger, C.; Ryoo, H.; Goldsmith, S. Dopamine D2 receptors in the hippocampus and amygdala in Alzheimer’s disease. Neurosci. Lett. 1993, 154, 171–174. [Google Scholar] [CrossRef]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Aourz, N.; Serruys, A.K.; Chabwine, J.N.; Balegamire, P.B.; Afrikanova, T.; Edrada-Ebel, R.; Grey, A.I.; Kamuhabwa, A.R.; Walrave, L.; Esguerra, C.V.; et al. Identification of GSK-3 as a Potential Therapeutic Entry Point for Epilepsy. ACS Chem. Neurosci. 2019, 10, 1992–2003. [Google Scholar] [CrossRef]
- Urbanska, M.; Kazmierska-Grebowska, P.; Kowalczyk, T.; Caban, B.; Nader, K.; Pijet, B.; Kalita, K.; Gozdz, A.; Devijver, H.; Lechat, B.; et al. GSK3beta activity alleviates epileptogenesis and limits GluA1 phosphorylation. EBioMedicine 2019, 39, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Matsuba, Y.; Yamazaki, N.; Hashimoto, S.; Saido, T.C. Calpain Activation in Alzheimer’s Model Mice Is an Artifact of APP and Presenilin Overexpression. J. Neurosci. 2016, 36, 9933–9936. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, R.; Jones, N.C.; Kwan, P. Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures. Int. J. Mol. Sci. 2020, 21, 3676. https://doi.org/10.3390/ijms21103676
Lin R, Jones NC, Kwan P. Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures. International Journal of Molecular Sciences. 2020; 21(10):3676. https://doi.org/10.3390/ijms21103676
Chicago/Turabian StyleLin, Runxuan, Nigel Charles Jones, and Patrick Kwan. 2020. "Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures" International Journal of Molecular Sciences 21, no. 10: 3676. https://doi.org/10.3390/ijms21103676
APA StyleLin, R., Jones, N. C., & Kwan, P. (2020). Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures. International Journal of Molecular Sciences, 21(10), 3676. https://doi.org/10.3390/ijms21103676