Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both?



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Seizures and the BBB
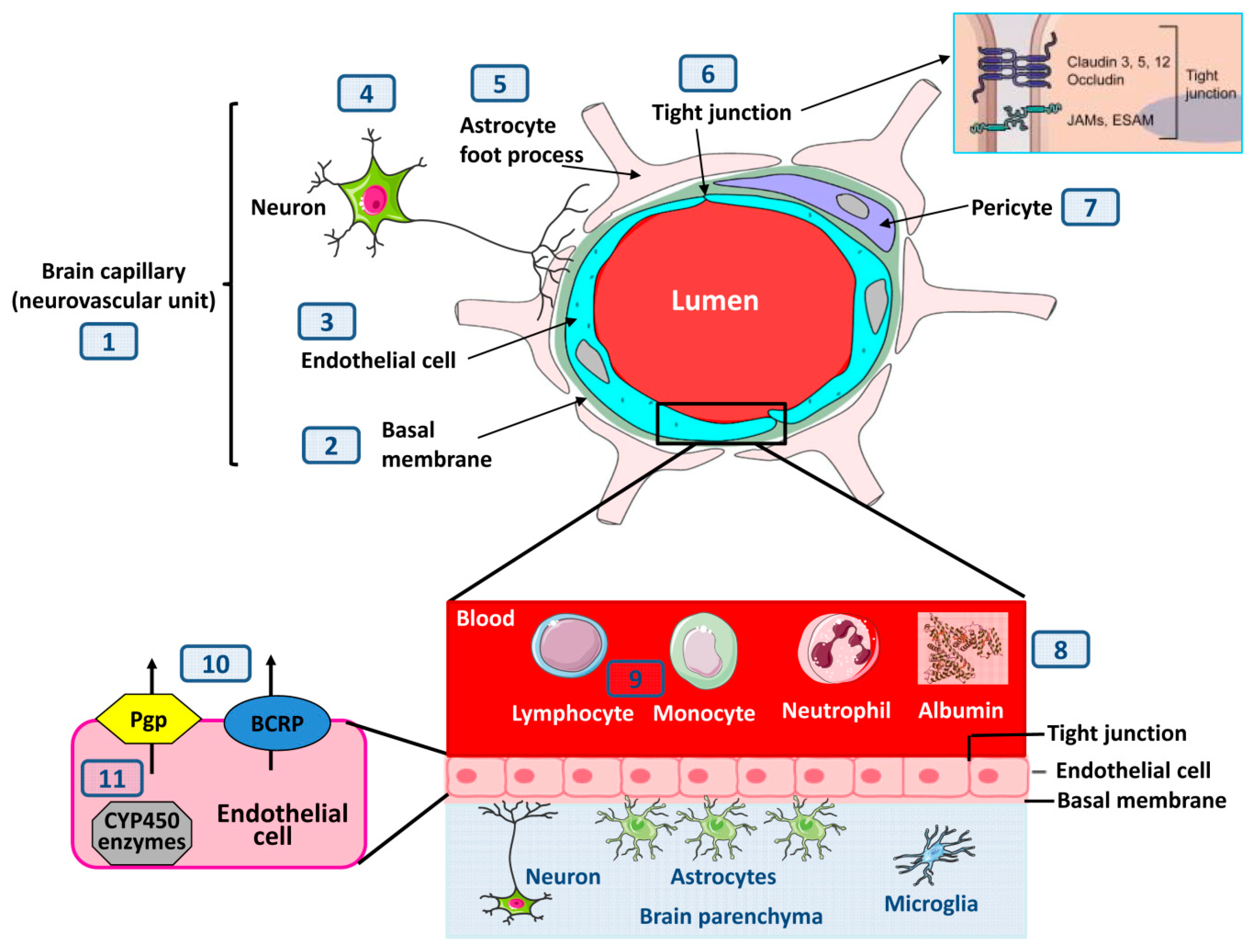
3. BBB Structural Alterations in Epilepsy
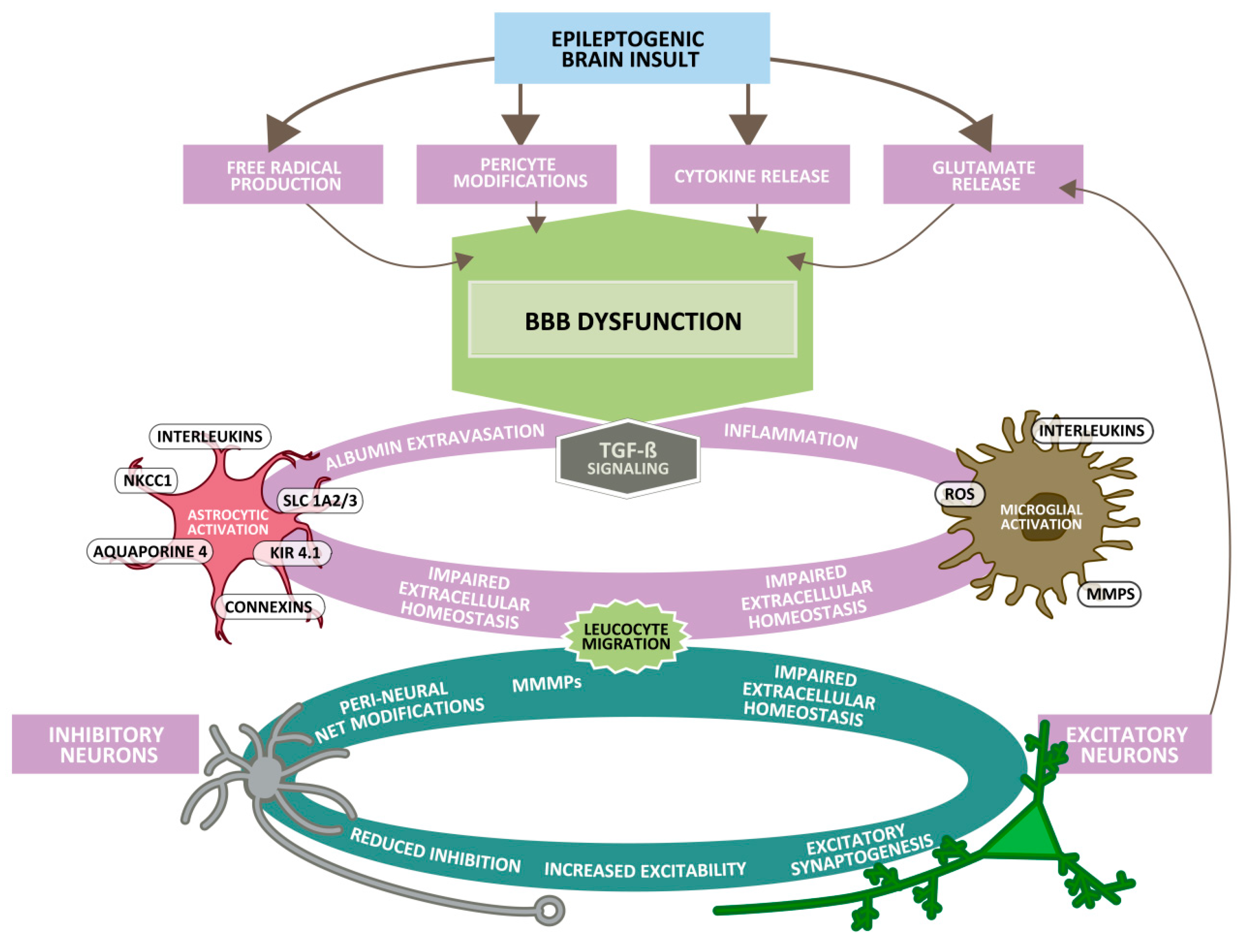
4. Why Do Epileptogenic Brain Insults and Seizures Alter the Morphology and Functionality of the BBB?
5. BBB Dysfunction May Alter Brain Uptake of Xenobiotics and Albumin
6. Increase in BBB Permeability Does Not always Lead to Seizures
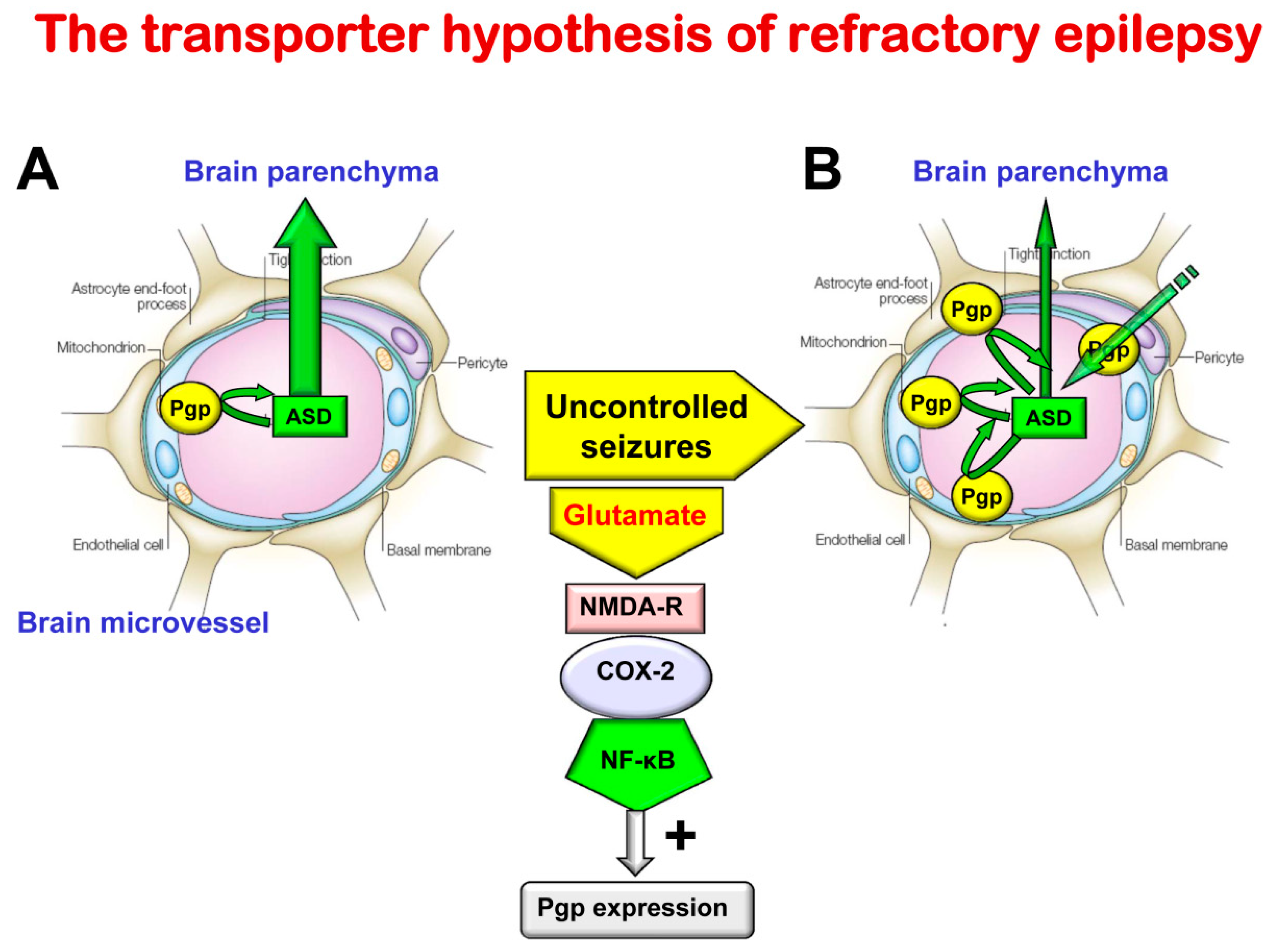
7. The Role of the BBB in Drug Resistance in Epilepsy
8. Impairment of Barrier Functions and the Invasion of Inflammatory Cells into the Brain
9. Impairment of Barrier Functions and its Role in the Development of Epilepsy
10. Overcoming the BBB in Epilepsy by Delivering Therapeutics to the Brain
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ehrlich, P. Das Sauerstoffbedürfnis des Organismus. Eine Farbanalytische Studie; Hirschwald-Verlag: Berlin, Germany, 1885. [Google Scholar]
- Stern, L. Le liquide céfalo-rachidien au point de vue de ses rapports avec la circulation sanguine et avec les éléments nerveux de l’axe cérébrospinal. Schweiz. Arch. Neurol. Psychiatr. 1921, 8, 215–232. [Google Scholar]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Serlin, Y.; Friedman, A.; Heinemann, U. The blood-brain barrier. In Homeostatic Control of Brain Function; Boison, D., Masino, S.A., Eds.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Seelig, A. The role of size and charge for blood-brain barrier permeation of drugs and fatty acids. J. Mol. Neurosci. 2007, 33, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Löscher, W.; Luna-Tortós, C.; Römermann, K.; Fedrowitz, M. Do ATP-binding cassette transporters cause pharmacoresistance in epilepsy? Problems and approaches in determining which antiepileptic drugs are affected. Curr. Pharm. Des. 2011, 17, 2808–2828. [Google Scholar] [CrossRef]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef]
- Frey, H.-H.; Löscher, W. Distribution of valproate across the interface between blood and cerebrospinal fluid. Neuropharmacology 1978, 17, 637–642. [Google Scholar] [CrossRef]
- Vijay, N.; Morris, M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, I.; Nyul-Toth, A.; Suciu, M.; Hermenean, A.; Krizbai, I.A. Heterogeneity of the blood-brain barrier. Tissue Barriers 2016, 4, e1143544. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog. Neurobiol. 2005, 76, 22–76. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef]
- Ghosh, C.; Puvenna, V.; Gonzalez-Martinez, J.; Janigro, D.; Marchi, N. Blood-brain barrier P450 enzymes and multidrug transporters in drug resistance: A synergistic role in neurological diseases. Curr. Drug Metab. 2011, 12, 742–749. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.F.; Leonhardt, H. Information on the hemato-encephalic barrier; cardiazole shock and barrier collapse. Arch. Psychiatr. Nervenkr. Z. Gesamte Neurol. Psychiatr. 1955, 193, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.F.; Leonhardt, H. A contribution to the pathological physiology of the blood-brain-barrier; megaphen stabilises the blood-brain-barrier. J. Comp. Neurol. 1956, 106, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; Aronica, E.; Gorter, J.A. Blood-brain barrier dysfunction, seizures and epilepsy. Semin. Cell Dev. Biol. 2015, 38, 26–34. [Google Scholar] [CrossRef]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef]
- Clarke, H.B.; Gabrielsen, T.O. Seizure induced disruption of blood-brain barrier demonstrated by CT. J. Comput. Assist. Tomogr. 1989, 13, 889–892. [Google Scholar] [CrossRef]
- Duncan, R.; Todd, N. Epilepsy and the blood-brain barrier. Br. J. Hosp. Med. 1991, 45, 32–34. [Google Scholar]
- Carvey, P.M. Drug Action in the Central Nervous System; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Kang, E.J.; Major, S.; Jorks, D.; Reiffurth, C.; Offenhauser, N.; Friedman, A.; Dreier, J.P. Blood-brain barrier opening to large molecules does not imply blood-brain barrier opening to small ions. Neurobiol. Dis. 2013, 52, 204–218. [Google Scholar] [CrossRef] [Green Version]
- Marchi, N.; Lerner-Natoli, M. Cerebrovascular remodeling and epilepsy. Neuroscientist 2013, 19, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Rigau, V.; Morin, M.; Rousset, M.C.; de Bock, F.; Lebrun, A.; Coubes, P.; Picot, M.C.; Baldy-Moulinier, M.; Bockaert, J.; Crespel, A.; et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain 2007, 130, 1942–1956. [Google Scholar] [CrossRef] [Green Version]
- Cornford, E.M.; Oldendorf, W.H. Epilepsy and the blood-brain barrier. Adv. Neurol. 1986, 44, 787–812. [Google Scholar]
- Cornford, E.M. Epilepsy and the blood brain barrier: Endothelial cell responses to seizures. Adv. Neurol. 1999, 79, 845–862. [Google Scholar]
- Giannoni, P.; Badaut, J.; Dargazanli, C.; De Maudave, A.F.; Klement, W.; Costalat, V.; Marchi, N. The pericyte-glia interface at the blood-brain barrier. Clin. Sci. 2018, 132, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Klement, W.; Garbelli, R.; Zub, E.; Rossini, L.; Tassi, L.; Girard, B.; Blaquiere, M.; Bertaso, F.; Perroy, J.; de Bock, F.; et al. Seizure progression and inflammatory mediators promote pericytosis and pericyte-microglia clustering at the cerebrovasculature. Neurobiol. Dis. 2018, 113, 70–81. [Google Scholar] [CrossRef]
- Jabs, R.; Seifert, G.; Steinhauser, C. Astrocytic function and its alteration in the epileptic brain. Epilepsia 2008, 49, 3–12. [Google Scholar] [CrossRef]
- David, Y.; Cacheaux, L.P.; Ivens, S.; Lapilover, E.; Heinemann, U.; Kaufer, D.; Friedman, A. Astrocytic dysfunction in epileptogenesis: Consequence of altered potassium and glutamate homeostasis? J. Neurosci. 2009, 29, 10588–10599. [Google Scholar] [CrossRef] [Green Version]
- Coulter, D.A.; Steinhäuser, C. Role of astrocytes in epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022434. [Google Scholar] [CrossRef] [Green Version]
- Rüber, T.; David, B.; Luchters, G.; Nass, R.D.; Friedman, A.; Surges, R.; Stocker, T.; Weber, B.; Deichmann, R.; Schlaug, G.; et al. Evidence for peri-ictal blood-brain barrier dysfunction in patients with epilepsy. Brain 2018, 141, 2952–2965. [Google Scholar] [CrossRef]
- Xhima, K.; Weber-Adrian, D.; Silburt, J. Glutamate Induces Blood-Brain Barrier Permeability through Activation of N-Methyl-D-Aspartate Receptors. J. Neurosci. 2016, 36, 12296–12298. [Google Scholar] [CrossRef] [Green Version]
- Rempe, R.G.; Hartz, A.M.S.; Soldner, E.L.B.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix Metalloproteinase-Mediated Blood-Brain Barrier Dysfunction in Epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef] [Green Version]
- Lischper, M.; Beuck, S.; Thanabalasundaram, G.; Pieper, C.; Galla, H.J. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010, 1326, 114–127. [Google Scholar] [CrossRef]
- Feng, Z.H.; Hao, J.; Ye, L.; Dayao, C.; Yan, N.; Yan, Y.; Chu, L.; Shi, F.D. Overexpression of mu-calpain in the anterior temporal neocortex of patients with intractable epilepsy correlates with clinicopathological characteristics. Seizure 2011, 20, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Prager, O.; Kamintsky, L.; Hasam-Henderson, L.A.; Schoknecht, K.; Wuntke, V.; Papageorgiou, I.; Swolinsky, J.; Muoio, V.; Bar-Klein, G.; Vazana, U.; et al. Seizure-induced microvascular injury is associated with impaired neurovascular coupling and blood-brain barrier dysfunction. Epilepsia 2019, 60, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Schoknecht, K.; David, Y.; Heinemann, U. The blood-brain barrier-gatekeeper to neuronal homeostasis: Clinical implications in the setting of stroke. Semin. Cell Dev. Biol. 2015, 38, 35–42. [Google Scholar] [CrossRef]
- Löscher, W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016, 126, 157–184. [Google Scholar] [CrossRef]
- Potschka, H.; Baltes, S.; Fedrowitz, M.; Löscher, W. Impact of seizure activity on free extracellular phenytoin concentrations in amygdala-kindled rats. Neuropharmacology 2011, 61, 909–917. [Google Scholar] [CrossRef]
- Potschka, H.; Löscher, W. A comparison of extracellular levels of phenytoin in amygdala and hippocampus of kindled and non-kindled rats. Neuroreport 2002, 13, 167–171. [Google Scholar] [CrossRef]
- Marchi, N.; Betto, G.; Fazio, V.; Fan, Q.; Ghosh, C.; Machado, A.; Janigro, D. Blood-brain barrier damage and brain penetration of antiepileptic drugs: Role of serum proteins and brain edema. Epilepsia 2009, 50, 664–677. [Google Scholar] [CrossRef]
- Salar, S.; Maslarova, A.; Lippmann, K.; Nichtweiss, J.; Weissberg, I.; Sheintuch, L.; Kunz, W.S.; Shorer, Z.; Friedman, A.; Heinemann, U. Blood-brain barrier dysfunction can contribute to pharmacoresistance of seizures. Epilepsia 2014, 55, 1255–1263. [Google Scholar] [CrossRef] [Green Version]
- Rambeck, B.; Jürgens, U.H.; May, T.W.; Pannek, H.W.; Behne, F.; Ebner, A.; Gorji, A.; Straub, H.; Speckmann, E.J.; Pohlmann-Eden, B.; et al. Comparison of brain extracellular fluid, brain tissue, cerebrospinal fluid, and serum concentrations of antiepileptic drugs measured intraoperatively in patients with intractable epilepsy. Epilepsia 2006, 47, 681–694. [Google Scholar] [CrossRef]
- Montagne, A.; Toga, A.W.; Zlokovic, B.V. Blood-Brain Barrier Permeability and Gadolinium: Benefits and Potential Pitfalls in Research. JAMA Neurol. 2016, 73, 13–14. [Google Scholar] [CrossRef]
- Van Vliet, E.A.; Dedeurwaerdere, S.; Cole, A.J.; Friedman, A.; Koepp, M.J.; Potschka, H.; Immonen, R.; Pitkanen, A.; Federico, P. WONOEP appraisal: Imaging biomarkers in epilepsy. Epilepsia 2017, 58, 315–330. [Google Scholar] [CrossRef]
- Elst, L.V.; Chapelle, F.; Laurent, S.; Muller, R.N. Stereospecific binding of MRI contrast agents to humen serum albumin: The case of Gd-(S)-EOB-DTPA (Eovist) and its (R) isomer. J. Biol. Inorg. Chem. 2001, 6, 196–200. [Google Scholar] [CrossRef]
- McDonald, R.J.; McDonald, J.S.; Kallmes, D.F.; Jentoft, M.E.; Paolini, M.A.; Murray, D.L.; Williamson, E.E.; Eckel, L.J. Gadolinium Deposition in Human Brain Tissues after Contrast-enhanced MR Imaging in Adult Patients without Intracranial Abnormalities. Radiology 2017, 285, 546–554. [Google Scholar] [CrossRef]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Cacheaux, L.P.; Kamintsky, L.; Prager, O.; Weissberg, I.; Schoknecht, K.; Cheng, P.; Kim, S.Y.; Wood, L.; Heinemann, U.; et al. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann. Neurol. 2014, 75, 864–875. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Lublinsky, S.; Kamintsky, L.; Noyman, I.; Veksler, R.; Dalipaj, H.; Senatorov, V.V., Jr.; Swissa, E.; Rosenbach, D.; Elazary, N.; et al. Imaging blood-brain barrier dysfunction as a biomarker for epileptogenesis. Brain 2017, 140, 1692–1705. [Google Scholar] [CrossRef]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blümcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J., Jr.; Forcelli, P.A.; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef]
- Abbott, N.J.; Khan, E.U.; Rollinson, C.M.S.; Reichel, A.; Janigro, D.; Dombrowski, S.M.; Dobbie, M.S.; Begley, D.J. Drug resistance in epilepsy: The role of the blood-brain barrier. In Mechanisms of Drug Resistance in Epilepsy. Lessons from Oncology; Ling, V., Ed.; Wiley: Chichester, UK, 2002; pp. 38–46. [Google Scholar]
- Löscher, W.; Schmidt, D. Modern antiepileptic drug development has failed to deliver: Ways out of the current dilemma. Epilepsia 2011, 52, 657–678. [Google Scholar] [CrossRef]
- Tishler, D.M.; Weinberg, K.T.; Hinton, D.R.; Barbaro, N.; Annett, G.M.; Raffel, C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia 1995, 36, 1–6. [Google Scholar] [CrossRef]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef]
- Miller, D.S.; Bauer, B.; Hartz, A.M.S. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve CNS pharmacotherapy. Pharm. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Bankstahl, J.P.; Hoffmann, K.; Bethmann, K.; Löscher, W. Glutamate is critically involved in seizure-induced overexpression of P-glycoprotein in the brain. Neuropharmacology 2008, 54, 1006–1016. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Pekcec, A.; Toellner, K.; Miller, D.S.; Potschka, H. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol. Pharmacol. 2008, 73, 1444–1453. [Google Scholar] [CrossRef]
- Zibell, G.; Unkruer, B.; Pekcec, A.; Hartz, A.M.; Bauer, B.; Miller, D.S.; Potschka, H. Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition. Neuropharmacology 2009, 56, 849–855. [Google Scholar] [CrossRef]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for antiepileptic drug discovery and development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef]
- Zhang, C.; Kwan, P.; Zuo, Z.; Baum, L. The transport of antiepileptic drugs by P-glycoprotein. Adv. Drug Deliv. Rev. 2012, 64, 930–942. [Google Scholar] [CrossRef]
- Feldmann, M.; Asselin, M.C.; Liu, J.; Wang, S.; McMahon, A.; Anton-Rodriguez, J.; Walker, M.; Symms, M.; Brown, G.; Hinz, R.; et al. P-glycoprotein expression and function in patients with temporal lobe epilepsy: A case-control study. Lancet Neurol. 2013, 12, 777–785. [Google Scholar] [CrossRef]
- Liu, J.Y.; Thom, M.; Catarino, C.B.; Martinian, L.; Figarella-Branger, D.; Bartolomei, F.; Koepp, M.; Sisodiya, S.M. Neuropathology of the blood-brain barrier and pharmaco-resistance in human epilepsy. Brain 2012, 135, 3115–3133. [Google Scholar] [CrossRef] [Green Version]
- Brandt, C.; Bethmann, K.; Gastens, A.M.; Löscher, W. The multidrug transporter hypothesis of drug resistance in epilepsy: Proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol. Dis. 2006, 24, 202–211. [Google Scholar] [CrossRef]
- Asadi-Pooya, A.A.; Razavizadegan, S.M.; Abdi-Ardekani, A.; Sperling, M.R. Adjunctive use of verapamil in patients with refractory temporal lobe epilepsy: A pilot study. Epilepsy Behav. 2013, 29, 150–154. [Google Scholar] [CrossRef]
- Borlot, F.; Wither, R.G.; Ali, A.; Wu, N.; Verocai, F.; Andrade, D.M. A pilot double-blind trial using verapamil as adjuvant therapy for refractory seizures. Epilepsy Res. 2014, 108, 1642–1651. [Google Scholar] [CrossRef]
- Narayanan, J.; Frech, R.; Walters, S.; Patel, V.; Frigerio, R.; Maraganore, D.M. Low dose verapamil as an adjunct therapy for medically refractory epilepsy—An open label pilot study. Epilepsy Res. 2016, 126, 197–200. [Google Scholar] [CrossRef]
- Chung, F.S.; Santiago, J.S.; Jesus, M.F.; Trinidad, C.V.; See, M.F. Disrupting P-glycoprotein function in clinical settings: What can we learn from the fundamental aspects of this transporter? Am. J. Cancer Res. 2016, 6, 1583–1598. [Google Scholar]
- Potschka, H. Modulating P-glycoprotein regulation: Future perspectives for pharmacoresistant epilepsies? Epilepsia 2010, 51, 1333–1347. [Google Scholar] [CrossRef]
- Römermann, K.; Helmer, R.; Löscher, W. The antiepileptic drug lamotrigine is a substrate of mouse and human breast cancer resistance protein (ABCG2). Neuropharmacology 2015, 93, 7–14. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Luna-Tortós, C.; Fedrowitz, M.; Löscher, W. Evaluation of transport of common antiepileptic drugs by human multidrug resistance-associated proteins (MRP1, 2 and 5) that are overexpressed in pharmacoresistant epilepsy. Neuropharmacology 2010, 58, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Vezzani, A. Pharmacological targeting of brain inflammation in epilepsy: Therapeutic perspectives from experimental and clinical studies. Epilepsia Open 2018, 3, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.M. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef] [Green Version]
- Imhof, B.A.; Dunon, D. Basic mechanism of leukocyte migration. Horm. Metab. Res. 1997, 29, 614–621. [Google Scholar] [CrossRef]
- Man, S.; Ubogu, E.E.; Ransohoff, R.M. Inflammatory cell migration into the central nervous system: A few new twists on an old tale. Brain Pathol. 2007, 17, 243–250. [Google Scholar] [CrossRef]
- Langer, H.F.; Chavakis, T. Leukocyte-endothelial interactions in inflammation. J. Cell Mol. Med. 2009, 13, 1211–1220. [Google Scholar] [CrossRef]
- Rossi, B.; Angiari, S.; Zenaro, E.; Budui, S.L.; Constantin, G. Vascular inflammation in central nervous system diseases: Adhesion receptors controlling leukocyte-endothelial interactions. J. Leukoc. Biol. 2011, 89, 539–556. [Google Scholar] [CrossRef] [PubMed]
- Odoardi, F.; Sie, C.; Streyl, K.; Ulaganathan, V.K.; Schlager, C.; Lodygin, D.; Heckelsmiller, K.; Nietfeld, W.; Ellwart, J.; Klinkert, W.E.; et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012, 488, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flugel, A.; Laman, J.D.; Weller, R.O. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016, 132, 317–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, N.; Flugel, A. Knocking at the brain’s door: Intravital two-photon imaging of autoreactive T cell interactions with CNS structures. Semin. Immunopathol. 2010, 32, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Pitsch, J.; Kuehn, J.C.; Gnatkovsky, V.; Muller, J.A.; van Loo, K.M.J.; De Curtis, M.; Vatter, H.; Schoch, S.; Elger, C.E.; Becker, A.J. Anti-epileptogenic and Anti-convulsive Effects of Fingolimod in Experimental Temporal Lobe Epilepsy. Mol. Neurobiol. 2019, 56, 1825–1840. [Google Scholar] [CrossRef]
- Fabene, P.F.; Navarro, M.G.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minogue, A.M. Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: Effects on cognition, learning and affective behaviour. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 15–18. [Google Scholar] [CrossRef]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef] [Green Version]
- Käufer, C.; Chatbar, C.; Bröer, S.; Waltl, I.; Luca, G.; Gerhauser, I.; Kalinke, U.; Löscher, W. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc. Natl. Acad. Sci. USA 2018, 115, E8929–E8938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Ivens, S.; Kaufer, D.; Flores, L.P.; Bechmann, I.; Zumsteg, D.; Tomkins, O.; Seiffert, E.; Heinemann, U.; Friedman, A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2007, 130, 535–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Friedman, A.; Dingledine, R.J. The role of inflammation in epileptogenesis. Neuropharmacology 2013, 69, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fieschi, C.; Lenzi, G.L.; Zanette, E.; Orzi, F.; Passero, S. Effects on EEG of the osmotic opening of the blood-brain barrier in rats. Life Sci. 1980, 27, 239–243. [Google Scholar] [CrossRef]
- Marchi, N.; Angelov, L.; Masaryk, T.; Fazio, V.; Granata, T.; Hernandez, N.; Hallene, K.; Diglaw, T.; Franic, L.; Najm, I.; et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 2007, 48, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Friedman, A.; Kaufer, D.; Heinemann, U. Blood-brain barrier breakdown-inducing astrocytic transformation: Novel targets for the prevention of epilepsy. Epilepsy Res. 2009, 85, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Senatorov, V.V., Jr.; Morrissey, C.S.; Lippmann, K.; Vazquez, O.; Milikovsky, D.Z.; Gu, F.; Parada, I.; Prince, D.A.; Becker, A.J.; et al. TGFbeta signaling is associated with changes in inflammatory gene expression and perineuronal net degradation around inhibitory neurons following various neurological insults. Sci. Rep. 2017, 7, 7711. [Google Scholar] [CrossRef]
- Weissberg, I.; Wood, L.; Kamintsky, L.; Vazquez, O.; Milikovsky, D.Z.; Alexander, A.; Oppenheim, H.; Ardizzone, C.; Becker, A.; Frigerio, F.; et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol. Dis. 2015, 78, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salar, S.; Lapilover, E.; Muller, J.; Hollnagel, J.O.; Lippmann, K.; Friedman, A.; Heinemann, U. Synaptic plasticity in area CA1 of rat hippocampal slices following intraventricular application of albumin. Neurobiol. Dis. 2016, 91, 155–165. [Google Scholar] [CrossRef]
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; He, J.C.; Wu, J.W.; Zhan, S.Q.; Zhang, G.L.; Wu, H.Q.; Zhang, R.; Gao, Z.; Ren, H.W. Losartan inhibits development of spontaneous recurrent seizures by preventing astrocyte activation and attenuating blood-brain barrier permeability following pilocarpine-induced status epilepticus. Brain Res. Bull. 2019, 149, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, O.; Shelef, I.; Kaizerman, I.; Eliushin, A.; Afawi, Z.; Misk, A.; Gidon, M.; Cohen, A.; Zumsteg, D.; Friedman, A. Blood-brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry 2008, 79, 774–777. [Google Scholar] [CrossRef] [Green Version]
- Pavlovsky, L.; Seiffert, E.; Heinemann, U.; Korn, A.; Golan, H.; Friedman, A. Persistent BBB disruption may underlie alpha interferon-induced seizures. J. Neurol. 2005, 252, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Bankstahl, M.; Breuer, H.; Leiter, I.; Markel, M.; Bascunana, P.; Michalski, D.; Bengel, F.M.; Löscher, W.; Meier, M.; Bankstahl, J.P.; et al. Blood-Brain Barrier Leakage during Early Epileptogenesis Is Associated with Rapid Remodeling of the Neurovascular Unit. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, R.; Chen, L.; Gotz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2019, in press. [Google Scholar] [CrossRef]
- Nilsen, K.E.; Cock, H.R. Focal treatment for refractory epilepsy: Hope for the future? Brain Res. Brain Res. Rev. 2004, 44, 141–153. [Google Scholar] [CrossRef]
- Rogawski, M.A. Convection-enhanced delivery in the treatment of epilepsy. Neurotherapeutics 2009, 6, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Van Dycke, A.; Raedt, R.; Vonck, K.; Boon, P. Local delivery strategies in epilepsy: A focus on adenosine. Seizure 2011, 20, 376–382. [Google Scholar] [CrossRef]
- Van Tienderen, G.S.; Berthel, M.; Yue, Z.; Cook, M.; Liu, X.; Beirne, S.; Wallace, G.G. Advanced fabrication approaches to controlled delivery systems for epilepsy treatment. Expert Opin. Drug Deliv. 2018, 15, 915–925. [Google Scholar] [CrossRef]
- Madhavan, D.; Mirowski, P.; Ludvig, N.; Carlson, C.; Doyle, W.; Devinsky, O.; Kuzniecky, R. Effects of subdural application of lidocaine in patients with focal epilepsy. Epilepsy Res. 2008, 78, 235–239. [Google Scholar] [CrossRef]
- Heiss, J.D.; Argersinger, D.P.; Theodore, W.H.; Butman, J.A.; Sato, S.; Khan, O.I. Convection-Enhanced Delivery of Muscimol in Patients with Drug-Resistant Epilepsy. Neurosurgery 2019, 85, E4–E15. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.J.; Murphy, M.A.; Bullus, K.; D’Souza, W.; Plummer, C.; Priest, E.; Williams, C.; Sharan, A.; Fisher, R.S.; Pincus, S.; et al. Anti-seizure therapy with a long-term, implanted intra-cerebroventricular delivery system in patients with drug-resistant epilepsy: A first-in-man study. In Proceedings of the American Epilepsy Society 73th Annual Meeting, Baltimore, MD, USA, 6–10 December 2019. [Google Scholar]
- Jasper, H.H. Physiopathological mechanisms of post-traumatic epilepsy. Epilepsia 1997, 11, 73–80. [Google Scholar] [CrossRef]
- Friedman, A.; Heinemann, U. Role of Blood-Brain Barrier Dysfunction in Epileptogenesis. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012; pp. 1–12. [Google Scholar]
- Friedman, A.; Bar-Klein, G.; Serlin, Y.; Parmet, Y.; Heinemann, U.; Kaufer, D. Should losartan be administered following brain injury? Expert Rev. Neurother. 2014, 14, 1365–1375. [Google Scholar] [CrossRef]
- Marchi, N.; Granata, T.; Ghosh, C.; Janigro, D. Blood-brain barrier dysfunction and epilepsy: Pathophysiologic role and therapeutic approaches. Epilepsia 2012, 53, 1877–1886. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Löscher, W.; Friedman, A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? Int. J. Mol. Sci. 2020, 21, 591. https://doi.org/10.3390/ijms21020591
Löscher W, Friedman A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? International Journal of Molecular Sciences. 2020; 21(2):591. https://doi.org/10.3390/ijms21020591
Chicago/Turabian StyleLöscher, Wolfgang, and Alon Friedman. 2020. "Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both?" International Journal of Molecular Sciences 21, no. 2: 591. https://doi.org/10.3390/ijms21020591
APA StyleLöscher, W., & Friedman, A. (2020). Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? International Journal of Molecular Sciences, 21(2), 591. https://doi.org/10.3390/ijms21020591