S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy



{kind=link}
Abstract
:1. Introduction
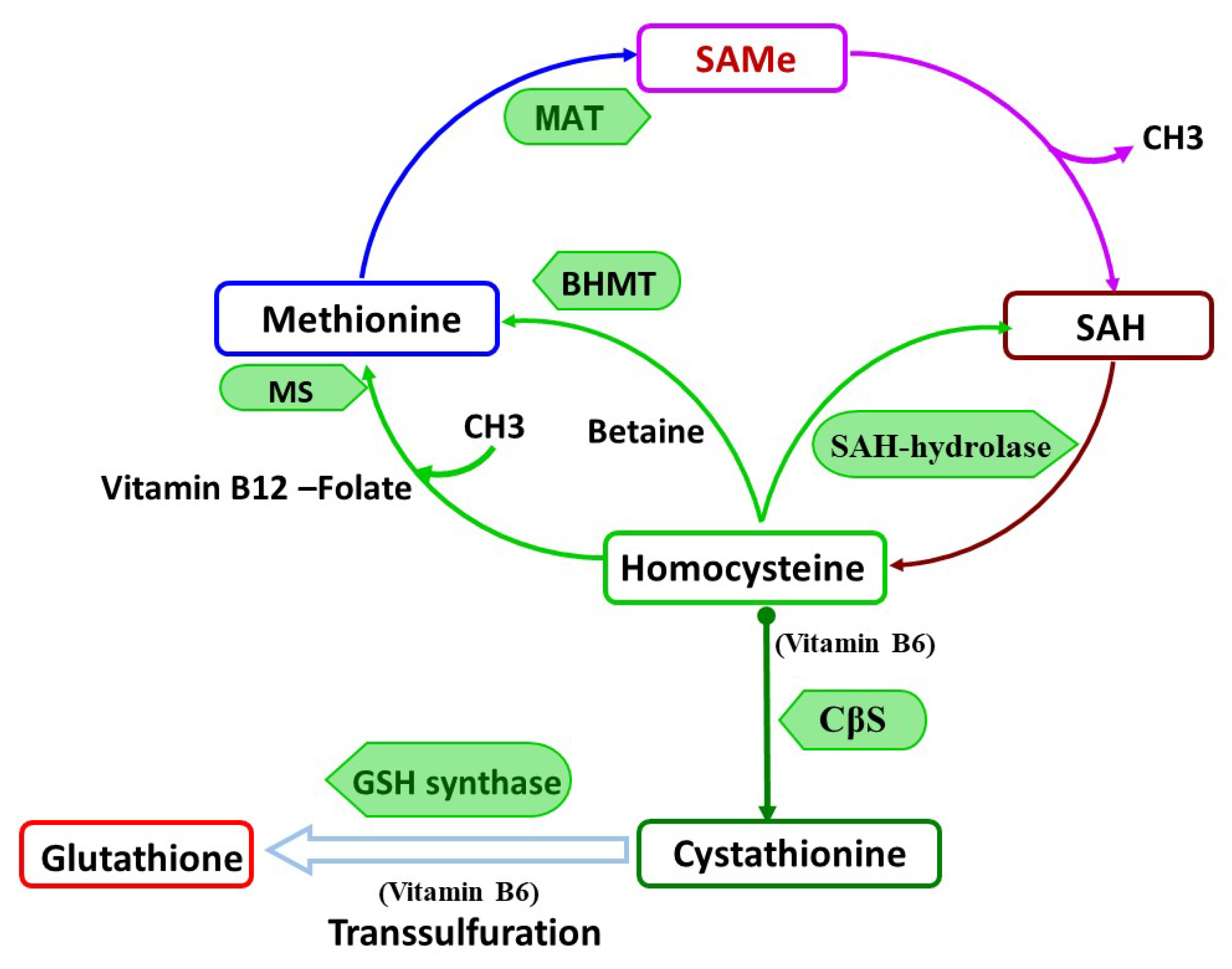
S-Adenosylmethionine: The Principal Physiological Methyl Donor
2. Overview of DNA Methylation and Chromatin Modification Machinery
2.1. Sensitive Periods for Epigenetic Changes
2.2. Differences and Similarities in Critical Periods between Rodents and Humans
2.3. Methyl Donors and Major Depression Disorder (MDD)
2.4. Homocysteine as a Possible Marker of SAMe Deficiency and Epigenetic Changes
2.5. Oxidative Stress in MDD, ASD, and Alzheimer Disease and the Effects of SAMe
3. Valproic Acid-Basic Pharmacology
Valproic Acid as a Histone Deacetylase Inhibitor (HDACI)
4. Hazardous Potential of VPA as HDACI
4.1. Teratogenicity
4.2. VPA and Neurological Diseases
5. The Treating Potential of VPA as HDACI
5.1. Alzheimer Disease (AD)
5.2. Bipolar Disease
5.3. Cancer
5.4. HIV
6. The Effects of SAMe in Pregnancy
7. Effects of SAMe and VPA in Pregnancy
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ASD | Autism spectrum disorder |
| 3-MHPG | 3-Methoxy-4-hydroxyphenylglycol |
| 5caC | 5-carboxyl-cytosine |
| 5fC | 5-formyl-cytosine |
| 5HIAA | 5-hydroxyindoleacetic acid |
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5methylcytosine |
| MECP2 | Methyl CPG binding protein 2 |
| ART | Antiretroviral therapy |
| ABCA1 | ATP-binding cassette transporter 1 |
| BACE1 | Beta-Secretase 1 |
| BDNF | Brain-derived neurotrophic factor |
| CPS1 | Carbamoyl phosphate synthetase 1 |
| CAT | Catalase |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CP | Critical period |
| d-AMPH | Dextroamphetamine |
| DNMTs | DNA methyltransferases |
| ERK | Extracellular signal-regulated kinase |
| Flt1 | Fms Related Receptor Tyrosine Kinase 1 |
| FST | Forced swim test |
| GABA | Gamma aminobutyric acid |
| GD | Gestational Day |
| UGT | Glucuronyl-transferase |
| GSH | Glutathione |
| GSH-Px | Glutathione peroxidase |
| GST | Glutathione-S-transferase |
| GSK-3 | Glycogen synthase kinase-3 |
| HAT | Histone acetyltransferases |
| HDACs | Histone deacetylases |
| HCY | Homocysteine |
| HVA | Homovanillic acid |
| HIV | Human immune deficiency virus |
| HPG axis | Hypothalamic–pituitary–gonadal axis |
| HIF1α | Hypoxia inducible factor alpha |
| IEG | Immediate–early gene |
| LSD | Lysine-specific demethylase |
| MARCKSL1 | Macrophage Myristoylated Alanine-Rich C Kinase Substrate |
| MDD | Major depression disorder |
| MBD | Methyl-binding domain |
| NGF | Nerve growth factor |
| NTD | Neural tube defects |
| NMDA | N-Methyl-D-Aspartate |
| NFkB | Nuclear factor kappa B |
| PLOG | Polymerase gamma gene |
| PSMS | Post-translational histone modifications |
| PSEN1 | Presenilin-1 |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SAH | S-adenosylhomocysteine |
| SAMe | S-adenosylmethionine |
| SAHH | SAH hydrolase |
| SCN1A | Sodium channel neuronal type 1 alpha |
| SOD | Superoxide dismutase |
| TET | Ten-eleven translocation |
| TdT | Terminal deoxynucleotidyl transferase |
| Thcy | Total HCY |
| TSA | Trichostatin A |
| TrkB | Tropomyosin-related kinase receptor B |
| VPA | Valproic acid |
| Vegf-a | Vascular endothelial growth factor a |
References
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. Prospects for the development of epigenetic drugs for CNS conditions. Nat. Rev. Drug Discov. 2015, 14, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Boil. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Bottiglieri, T. S-Adenosyl-L-methionine (SAMe): from the bench to the bedside--molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 2002, 76, 1151S–1157S. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.W.; Shukitt-Hale, B.; Villalobos-Molina, R.; Nadeau, M.R.; Selhub, J.; Joseph, J.A. Effect of L-Dopa and the catechol-O-methyltransferase inhibitor Ro 41-0960 on sulfur amino acid metabolites in rats. Clin. Neuropharmacol. 1997, 20, 55–66. [Google Scholar] [CrossRef]
- Cheng, H.; Gomes-Trolin, C.; Aquilonius, S.-M.; Steinberg, A.; Löfberg, C.; Ekblom, J.; Oreland, L. Levels ofl-MethionineS-Adenosyltranferase Activity in Erythrocytes and Concentrations ofS-Adenosylmethionine andS-Adenosylhomocysteine in Whole Blood of Patients with Parkinson’s Disease. Exp. Neurol. 1997, 145, 580–585. [Google Scholar] [CrossRef]
- Surtees, R.; Hyland, K. L-3,4-dihydroxyphenylalanine (levodopa) lowers central nervous system S-adenosylmethionine concentrations in humans. J. Neurol. Neurosurg. Psychiatry 1990, 53, 569–572. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Álvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Ragnarsson, L.; Mortensen, M.; Dodd, P.R.; Lewis, R.J. Spermine modulation of the glutamate NMDA receptor is differentially responsive to conantokins in normal and Alzheimer’s disease human cerebral cortex. J. Neurochem. 2002, 81, 765–779. [Google Scholar] [CrossRef]
- Mudd, S.; Poole, J.R. Labile methyl balances for normal humans on various dietary regimens. Metabolism 1975, 24, 721–735. [Google Scholar] [CrossRef]
- Chen, Z.-X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Boil. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, A.J.; Stathaki, E.; Migliavacca, E.; Brahmachary, M.; Montgomery, S.B.; Dupré, Y.; Antonarakis, S.E. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011, 21, 1592–1600. [Google Scholar] [CrossRef] [Green Version]
- Hanson, M.A.; Gluckman, P.D. Developmental origins of health and disease--global public health implications. Best Pract. Res. Clin. Obstet. Gynaecol. 2015, 29, 24–31. [Google Scholar] [CrossRef]
- Kuijper, B.; Hanson, M.A.; Vitikainen, E.I.K.; Marshall, H.H.; Ozanne, S.E.; Cant, M.A. Developing differences: early-life effects and evolutionary medicine. Philos. Trans. R. Soc. B Boil. Sci. 2019, 374, 20190039. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Jeltsch, A.; Jurkowska, R.Z. Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Res. 2016, 44, 8556–8575. [Google Scholar] [CrossRef] [Green Version]
- Jeltsch, A. Reading and writing DNA methylation. Nat. Struct. Mol. Boil. 2008, 15, 1003–1004. [Google Scholar] [CrossRef]
- Cheng, X.; Blumenthal, R. Mammalian DNA Methyltransferases: A Structural Perspective. Structure 2008, 16, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef] [PubMed]
- Ausio, J.; De Paz, A.M.; Esteller, M. MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 2014, 20, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. Methylation meets acetylation. Nature 1998, 393, 311–312. [Google Scholar] [CrossRef]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Molina-Serrano, D.; Kyriakou, D.; Kirmizis, A. Histone Modifications as an Intersection Between Diet and Longevity. Front. Genet. 2019, 10, 192. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Tan, L.; Shi, Y.G. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Rao, A.; Ko, M. TET family dioxygenases and DNA demethylation in stem cells and cancers. Exp. Mol. Med. 2017, 49, e323. [Google Scholar] [CrossRef] [Green Version]
- Nagy, C.; Turecki, G. Sensitive periods in epigenetics: Bringing us closer to complex behavioral phenotypes. Epigenomics 2012, 4, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell 2014, 14, 710–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.; Meletis, K.; Fu, N.; Jhaveri, S.; Jaenisch, R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn. 2007, 236, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.; Faull, K.F.; Lyko, F.; et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 2014, 29, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Roost, M.S.; Slieker, R.C.; Bialecka, M.; Van Iperen, L.; Fernandes, M.M.G.; He, N.; Suchiman, H.E.; Szuhai, K.; Carlotti, F.; De Koning, E.J.P.; et al. DNA methylation and transcriptional trajectories during human development and reprogramming of isogenic pluripotent stem cells. Nat. Commun. 2017, 8, 908. [Google Scholar] [CrossRef] [PubMed]
- Reizel, Y.; Sabag, O.; Skversky, Y.; Spiro, A.; Steinberg, B.; Bernstein, D.; Wang, A.; Kieckhaefer, J.; Li, C.; Pikarsky, E.; et al. Postnatal DNA demethylation and its role in tissue maturation. Nat. Commun. 2018, 9, 2040. [Google Scholar] [CrossRef] [PubMed]
- Almstrup, K.; Johansen, M.L.; Busch, A.S.; Hagen, C.P.; Nielsen, J.E.; Petersen, J.H.; Juul, A. Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood. Sci. Rep. 2016, 6, 28657. [Google Scholar] [CrossRef]
- Yuan, X.; Li, Z.; Ye, S.; Chen, Z.; Huang, S.; Zhong, Y.; Zhang, H.; Li, J.; Zhang, Z. Genome-wide DNA methylation analysis of pituitaries during the initiation of puberty in gilts. PLoS ONE 2019, 14, e0212630. [Google Scholar] [CrossRef]
- A Bayer, S.; Altman, J.; Russo, R.J.; Zhang, X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. NeuroToxicology 1993, 14, 83–144. [Google Scholar]
- Rice, D.; Barone, S.J. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. 3), 511–533. [Google Scholar]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, B.L.; Babb, S.M.; Ravichandran, C.; Cohen, B.M. Oral SAMe in Persistent Treatment-Refractory Bipolar Depression. J. Clin. Psychopharmacol. 2014, 34, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A. S-Adenosyl Methionine (SAMe) for Depression in Adults. Issues Ment. Heal. Nurs. 2019, 40, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. Epigenomics of Major Depressive Disorders and Schizophrenia: Early Life Decides. Int. J. Mol. Sci. 2017, 18, 1711. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Orsolini, L.; Serroni, N.; Girinelli, G.; Iasevoli, F.; Tomasetti, C.; De Bartolomeis, A.; Mazza, M.; Valchera, A.; Fornaro, M.; et al. A comprehensive review on the efficacy of S-Adenosyl-L-methionine in Major Depressive Disorder. CNS Neurol. Disord.-Drug Targets 2016, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.; Danneskiold-Samsøe, B.; Andersen, R.B. Oral S-adenosylmethionine in Primary Fibromyalgia. Double-blind Clinical Evaluation. Scand. J. Rheumatol. 1991, 20, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Nowak, H. A new medical approach to the treatment of osteoarthritis. Report of an open phase IV study with ademetionine (Gumbaral). Am. J. Med. 1987, 83, 84–88. [Google Scholar] [CrossRef]
- Friedel, H.A.; Goa, K.L.; Benfield, P. S-Adenosyl-L-Methionine. Drugs 1989, 38, 389–416. [Google Scholar] [CrossRef]
- Fernández, L.; Perez, V.; Muñoz, M.; Corpa, J.M.; Abad, M.; Carbajo, M.T. Effects of S-adenosylmethionine on hepatic regeneration after partial hepatectomy in the rat. J. Physiol. Biochem. 2003, 59, 63–64. [Google Scholar] [CrossRef]
- Purohit, V.; Abdelmalek, M.F.; Barve, S.; Benevenga, N.J.; Halsted, C.H.; Kaplowitz, N.; Kharbanda, K.K.; Liu, Q.-Y.; Lu, S.C.; McClain, C.J.; et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: summary of a symposium. Am. J. Clin. Nutr. 2007, 86, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Papakostas, G.I.; Mischoulon, D.; Shyu, I.; Alpert, J.E.; Fava, M. S-Adenosyl Methionine (SAMe) Augmentation of Serotonin Reuptake Inhibitors for Antidepressant Nonresponders With Major Depressive Disorder: A Double-Blind, Randomized Clinical Trial. Am. J. Psychiatry 2010, 167, 942–948. [Google Scholar] [CrossRef]
- Sarris, J.; Byrne, G.J.; Bousman, C.A.; Stough, C.; Murphy, J.; Macdonald, P.; Adams, L.; Nazareth, S.; Oliver, G.; Cribb, L.; et al. Adjunctive S-adenosylmethionine (SAMe) in treating non-remittent major depressive disorder: An 8-week double-blind, randomized, controlled trial. Eur. Neuropsychopharmacol. 2018, 28, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Mischoulon, D.; Fava, M. Role of S-adenosyl-L-methionine in the treatment of depression: A review of the evidence. Am. J. Clin. Nutr. 2002, 76, 1158S–1161S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, C.; Jackson, N.L.; Day, J.J.; Clinton, S.M. Genetic predisposition to high anxiety- and depression-like behavior coincides with diminished DNA methylation in the adult rat amygdala. Behav. Brain Res. 2016, 320, 165–178. [Google Scholar] [CrossRef] [Green Version]
- McCoy, C.; Jackson, N.L.; Brewer, R.L.; Moughnyeh, M.M.; Smith, D.L.; Clinton, S.M. A paternal methyl donor depleted diet leads to increased anxiety- and depression-like behavior in adult rat offspring. Biosci. Rep. 2018, 38, 20180730. [Google Scholar] [CrossRef] [Green Version]
- Darby, M.; Yolken, R.H.; Sabunciyan, S. Consistently altered expression of gene sets in postmortem brains of individuals with major psychiatric disorders. Transl. Psychiatry 2016, 6, e890. [Google Scholar] [CrossRef] [PubMed]
- Saunderson, E.A.; Spiers, H.; Mifsud, K.R.; Gutierrez-Mecinas, M.; Trollope, A.F.; Shaikh, A.; Mill, J.; Reul, J.M.H.M. Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2016, 113, 4830–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2015, 16, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Fancourt, D.; Steptoe, A. The longitudinal relationship between changes in wellbeing and inflammatory markers: Are associations independent of depression? Brain, Behav. Immun. 2019, 83, 146–152. [Google Scholar] [CrossRef]
- Song, Z.; Uriarte, S.; Sahoo, R.; Chen, T.; Barve, S.; Hill, D.; McClain, C.J. S-adenosylmethionine (SAMe) modulates interleukin-10 and interleukin-6, but not TNF, production via the adenosine (A2) receptor. Biochim. Biophys. Acta (BBA) Bioenergy 2005, 1743, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Gobejishvili, L.; Avila, D.V.; Barker, D.F.; Ghare, S.; Henderson, D.; Brock, G.N.; Kirpich, I.A.; Joshi-Barve, S.; Mokshagundam, S.P.L.; McClain, C.J.; et al. S-adenosylmethionine decreases lipopolysaccharide-induced phosphodiesterase 4B2 and attenuates tumor necrosis factor expression via cAMP/protein kinase A pathway. J. Pharmacol. Exp. Ther. 2011, 337, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Ara, A.; Xia, M.; Ramani, K.; Mato, J.M.; Lu, S.C. S-adenosylmethionine inhibits lipopolysaccharide-induced gene expression via modulation of histone methylation. Hepatology 2008, 47, 1655–1666. [Google Scholar] [CrossRef] [Green Version]
- Folstein, M.; Liu, T.; Peter, I.; Buel, J.; Arsenault, L.; Scott, T.; Qiu, W.W. The homocysteine hypothesis of depression. Am. J. Psychiatry 2007, 164, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Homocysteine Is a Protein Amino Acid in Humans. J. Boil. Chem. 2002, 277, 30425–30428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halasova, E.; Lehotsky, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef] [PubMed]
- Stanger, O.; Fowler, B.; Piertzik, K.; Huemer, M.; Haschke-Becher, E.; Semmler, A.; Lorenzl, S.; Linnebank, M. Homocysteine, folate and vitamin B12in neuropsychiatric diseases: review and treatment recommendations. Expert Rev. Neurother. 2009, 9, 1393–1412. [Google Scholar] [CrossRef] [Green Version]
- Bottiglieri, T.; Laundy, M.; Crellin, R.; Toone, B.K.; Carney, M.W.P.; Reynolds, E.H. Homocysteine, folate, methylation, and monoamine metabolism in depression. J. Neurol. Neurosurg. Psychiatry 2000, 69, 228–232. [Google Scholar] [CrossRef]
- Morris, M.S.; Fava, M.; Jacques, P.F.; Selhub, J.; Rosenberg, I.H. Depression and folate status in the US Population. Psychother. Psychosom. 2003, 72, 80–87. [Google Scholar] [CrossRef]
- Hintikka, J.; Tolmunen, T.; Tanskanen, A.; Viinamäki, H. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry 2003, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Ornoy, A. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod. Toxicol 2007, 24, 31–41. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W.J.H. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Pandya, C.D.; Howell, K.R.; Pillai, A. Antioxidants as potential therapeutics for neuropsychiatric disorders. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2012, 46, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarandöl, A.; Sarandol, E.; Eker, S.S.; Erdinc, S.; Vatansever, E.; Kirli, S. Major depressive disorder is accompanied with oxidative stress: short-term antidepressant treatment does not alter oxidative–antioxidative systems. Hum. Psychopharmacol. Clin. Exp. 2007, 22, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Krause, K.-H. NOX Enzymes in the Central Nervous System: From Signaling to Disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Cumurcu, B.E.; Ozyurt, H.; Etikan, I.; Demir, S.; Karlidag, R. Total antioxidant capacity and total oxidant status in patients with major depression: Impact of antidepressant treatment. Psychiatry Clin. Neurosci. 2009, 63, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Vavakova, M.; Ďuračková, Z.; Trebatická, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxid. Med. Cell. Longev. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Fišar, Z.; Medina, M.; Scapagnini, G.; Nowak, G.; Berk, M. New drug targets in depression: inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates—Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology 2012, 20, 127–150. [Google Scholar] [CrossRef]
- Shelton, R.; Claiborne, J.; Sidoryk-Wegrzynowicz, M.; Reddy, R.; Aschner, M.; Lewis, D.A.; Mirnics, K. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol. Psychiatry 2010, 16, 751–762. [Google Scholar] [CrossRef]
- Rybka, J.; Kędziora-Kornatowska, K.; Banaś-Leżańska, P.; Majsterek, I.; Carvalho, L.A.; Cattaneo, A.; Anacker, C.; Kędziora, J. Interplay between the pro-oxidant and antioxidant systems and proinflammatory cytokine levels, in relation to iron metabolism and the erythron in depression. Free Radic. Biol. Med. 2013, 63, 187–194. [Google Scholar] [CrossRef]
- Liu, T.; Zhong, S.; Liao, X.; Chen, J.; He, T.; Lai, S.; Jia, Y. A Meta-Analysis of Oxidative Stress Markers in Depression. PLoS ONE 2015, 10, e0138904. [Google Scholar] [CrossRef]
- Bilici, M.; Efe, H.; Köroğlu, M.; Uydu, H.A.; Bekaroğlu, M.; Değer, O. Antioxidative enzyme activities and lipid peroxidation in major depression: alterations by antidepressant treatments. J. Affect. Disord. 2001, 64, 43–51. [Google Scholar] [CrossRef]
- Milaneschi, Y.; Cesari, M.; Simonsick, E.M.; Vogelzangs, N.; Kanaya, A.M.; Yaffe, K.; Patrignani, P.; Metti, A.; Kritchevsky, S.B.; Pahor, M.; et al. Lipid Peroxidation and Depressed Mood in Community-Dwelling Older Men and Women. PLoS ONE 2013, 8, e65406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.-F.; Qi, X.-R.; Zhao, J.; Balesar, R.; Bao, A.-M.; Swaab, D.F. Decreased NOS1 Expression in the Anterior Cingulate Cortex in Depression. Cereb. Cortex 2012, 23, 2956–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.; Baral, M.; Geis, E.; Mitchell, J.; Ingram, J.; Hensley, A.; Zappia, I.; Newmark, S.; Gehn, E.; Rubin, R.A.; et al. The Severity of Autism Is Associated with Toxic Metal Body Burden and Red Blood Cell Glutathione Levels. J. Toxicol. 2009, 2009, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezzo, A.; Visconti, P.; Abruzzo, P.M.; Bolotta, A.; Ferreri, C.; Gobbi, G.; Malisardi, G.; Manfredini, S.; Marini, M.; Nanetti, L.; et al. Oxidative Stress and Erythrocyte Membrane Alterations in Children with Autism: Correlation with Clinical Features. PLoS ONE 2013, 8, e66418. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; E Frye, R.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Rios, P.G.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; DiMauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef]
- Huang, Z.; Zhong, X.-M.; Li, Z.-Y.; Feng, C.-R.; Pan, A.-J.; Mao, Q.-Q. Curcumin reverses corticosterone-induced depressive-like behavior and decrease in brain BDNF levels in rats. Neurosci. Lett. 2011, 493, 145–148. [Google Scholar] [CrossRef]
- Kulkarni, S.K.; Bhutani, M.K.; Bishnoi, M. Antidepressant activity of curcumin: involvement of serotonin and dopamine system. Psychopharmacol. 2008, 201, 435–442. [Google Scholar] [CrossRef]
- Bouayed, J.; Rammal, H.; Dicko, A.; Younos, C.; Soulimani, R. Chlorogenic acid, a polyphenol from Prunus domestica (Mirabelle), with coupled anxiolytic and antioxidant effects. J. Neurol. Sci. 2007, 262, 77–84. [Google Scholar] [CrossRef]
- Banji, D.; Banji, O.J.; Abbagoni, S.; Hayath, S.; Kambam, S.; Chiluka, V.L. Amelioration of behavioral aberrations and oxidative markers by green tea extract in valproate induced autism in animals. Brain Res. 2011, 1410, 141–151. [Google Scholar] [CrossRef]
- Abdulla, A.; Zhao, X.; Yang, F. Natural Polyphenols Inhibit Lysine-Specific Demethylase-1 in vitro. J. Biochem. Pharmacol. Res. 2013, 1, 56–63. [Google Scholar] [PubMed]
- Pragnya, B.; Kameshwari, J.; Veeresh, B. Ameliorating effect of piperine on behavioral abnormalities and oxidative markers in sodium valproate induced autism in BALB/C mice. Behav. Brain Res. 2014, 270, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Bosker, F.J.; Li, J.; Schoevers, R.A. N-acetylcysteine as add-on to antidepressant medication in therapy refractory major depressive disorder patients with increased inflammatory activity: study protocol of a double-blind randomized placebo-controlled trial. BMC Psychiatry 2018, 18, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasebe, K.; Gray, L.; Bortolasci, C.C.; Panizzutti, B.; Mohebbi, M.; Kidnapillai, S.; Spolding, B.; Walder, K.; Berk, M.; Malhi, G.; et al. Adjunctive N-acetylcysteine in depression: exploration of interleukin-6, C-reactive protein and brain-derived neurotrophic factor. Acta Neuropsychiatr. 2017, 29, 337–346. [Google Scholar] [CrossRef]
- Berk, M.; Dean, O.; Cotton, S.M.; Jeavons, S.; Tanious, M.; Kohlmann, K.; Hewitt, K.; Moss, K.; Allwang, C.; Schapkaitz, I.; et al. The Efficacy of Adjunctive N -Acetylcysteine in Major Depressive Disorder. J. Clin. Psychiatry 2014, 75, 628–636. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I. Hepatoprotective effects of S-adenosyl-L-methionine against alcohol- and cytochrome P450 2E1-induced liver injury. World J. Gastroenterol. 2010, 16, 1366–1376. [Google Scholar] [CrossRef]
- Lieber, C.S.; Casini, A.; DeCarli, L.M.; Kim, C.-I.; Lowe, N.; Sasaki, R.; Leo, M.A. S-Adenosyl-L-methionine attenuates alcohol-induced liver injury in the baboon. Hepatology 1990, 11, 165–172. [Google Scholar] [CrossRef]
- Guo, T.; Chang, L.; Xiao, Y.; Liu, Q. S-Adenosyl-L-Methionine for the Treatment of Chronic Liver Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0122124. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Ricceri, L.; Cavallaro, R.A.; Isopi, E.; Mangia, F.; Fiorenza, M.T.; Scarpa, S. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol. Aging 2012, 33, 1482.e1–1482.e16. [Google Scholar] [CrossRef]
- Cavallaro, R.A.; Nicolia, V.; Fiorenza, M.T.; Scarpa, S.; Fuso, A. S-Adenosylmethionine and Superoxide Dismutase 1 Synergistically Counteract Alzheimer’s Disease Features Progression in TgCRND8 Mice. Antioxidants 2017, 6, 76. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cui, J.; Fang, C.; Liu, M.; Min, G.; Li, L. S-Adenosylmethionine Attenuates Oxidative Stress and Neuroinflammation Induced by Amyloid-β Through Modulation of Glutathione Metabolism. J. Alzheimer’s Dis. 2017, 58, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, R.A.; Fuso, A.; Nicolia, V.; Scarpa, S. S-Adenosylmethionine Prevents Oxidative Stress and Modulates Glutathione Metabolism in TgCRND8 Mice Fed a B-Vitamin Deficient Diet. J. Alzheimer’s Dis. 2010, 20, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Persichilli, S.; Gervasoni, J.; Di Napoli, A.; Fuso, A.; Nicolia, V.; Giardina, B.; Scarpa, S.; Desiderio, C.; Cavallaro, R.A. Plasma Thiols Levels in Alzheimer’s Disease Mice under Diet-Induced Hyperhomocysteinemia: Effect of S-Adenosylmethionine and Superoxide-Dismutase Supplementation. J. Alzheimer’s Dis. 2015, 44, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef]
- Christensen, J.; Grønborg, T.K.; Sørensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013, 309, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Bialer, M. Why are antiepileptic drugs used for nonepileptic conditions? Epilepsia 2012, 53, 26–33. [Google Scholar] [CrossRef]
- Johannessen, C.U.; Johannessen, S.I. Valproate: Past, Present, and Future. CNS Drug Rev. 2006, 9, 199–216. [Google Scholar] [CrossRef]
- Ghodke, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Ma, R.; Liao, Q.; Tian, J.; Li, L.; Han, Y.; Zhang, X.; Ren, X.; Song, G.; Guo, Q.; et al. Upregulation of CD54 and downregulation of HLA-ABC contribute to the novel enhancement of the susceptibility of HL-60 cells to NK cell-mediated cytolysis induced by ATRA plus VPA. Oncol. Rep. 2016, 37, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Oniki, K.; Nishimura, M.; Ogusu, N.; Shimomasuda, M.; Ono, T.; Matsuda, K.; Yasui-Furukori, N.; Nakagawa, K.; Ishitsu, T.; et al. Determination of the Optimal Concentration of Valproic Acid in Patients with Epilepsy: A Population Pharmacokinetic-Pharmacodynamic Analysis. PLoS ONE 2015, 10, e0141266. [Google Scholar] [CrossRef]
- Phiel, C.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J. Boil. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terbach, N.; Williams, R.S. Structure–function studies for the panacea, valproic acid. Biochem. Soc. Trans. 2009, 37, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sixto-López, Y.; Bello, M.; Correa-Basurto, J. Exploring the inhibitory activity of valproic acid against the HDAC family using an MMGBSA approach. J. Comput. Aided Mol. Des. 2020. [Google Scholar] [CrossRef]
- Chanda, S.; Ang, C.E.; Lee, Q.Y.; Ghebrial, M.; Haag, D.; Shibuya, Y.; Wernig, M.; Südhof, T.C. Direct Reprogramming of Human Neurons Identifies MARCKSL1 as a Pathogenic Mediator of Valproic Acid-Induced Teratogenicity. Cell Stem Cell 2019, 25, 103–119.e6. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, S.; Takuma, K.; Hara, Y.; Maeda, Y.; Ago, Y.; Matsuda, T. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int. J. Neuropsychopharmacol. 2013, 16, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldrich, R.X.; Leanage, G.; She, D.; Dolan-Evans, E.; Nelson, M.; Reza, N.; Reutens, D. Inhibition of histone deacetylase in utero causes sociability deficits in postnatal mice. Behav. Brain Res. 2013, 257, 253–264. [Google Scholar] [CrossRef]
- Qing, H.; He, G.; Ly, P.T.T.; Fox, C.J.; Staufenbiel, M.; Cai, F.; Zhang, Z.; Wei, S.; Sun, X.; Chen, C.-H.; et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J. Exp. Med. 2008, 205, 2781–2789. [Google Scholar] [CrossRef]
- Noh, H.; Seo, H. Age-dependent effects of valproic acid in Alzheimer’s disease (AD) mice are associated with nerve growth factor (NGF) regulation. Neuroscience 2014, 266, 255–265. [Google Scholar] [CrossRef]
- Dedoni, S.; Marras, L.; Olianas, M.C.; Ingianni, A.; Onali, P. Downregulation of TrkB Expression and Signaling by Valproic Acid and Other Histone Deacetylase Inhibitors. J. Pharmacol. Exp. Ther. 2019, 370, 490–503. [Google Scholar] [CrossRef]
- Kavoosi, F.; Sanaei, M. Effect of 5-aza-2’-deoxycytidine in comparison to valproic acid and trichostatin a on histone deacetylase 1, dna methyltransferase 1, and cip/kip family (p21, p27, and p57) genes expression, cell growth inhibition, and apoptosis induction in colon cancer sw480 cell line. Adv. Biomed. Res. 2019, 8, 52. [Google Scholar] [CrossRef]
- Chen, J.-H.; Zheng, Y.-L.; Xu, C.; Gu, L.-Z.; Ding, Z.-L.; Qin, L.; Wang, Y.; Fu, R.; Wan, Y.-F.; Hu, C.-P. Valproic acid (VPA) enhances cisplatin sensitivity of non-small cell lung cancer cells via HDAC2 mediated down regulation of ABCA1. Boil. Chem. 2017, 398, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Montani, M.S.G.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Crosby, B.; Deas, C. Repurposing medications for use in treating HIV infection: A focus on valproic acid as a latency-reversing agent. J. Clin. Pharm. Ther. 2018, 43, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Ribalta, J.; Reyes, H.; Gonzalez, M.C.; Iglesias, J.; Arrese, M.; Poniachik, J.; Molina, C.; Segovia, N. S-Adenosyl-L-methionine in the treatment of patients with intrahepatic cholestasis of pregnancy: A randomized, double-blind, placebo-controlled study with negative results. Hepatology 1991, 13, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Ubeda-Martín, N.; Alonso-Aperte, E.; Achón, M.; Varela-Moreiras, G.; Puerta, J.; De Miguelsanz, J.P. [Morphological changes induced by valproate and its administration concomitant with folinic acid or S-adenosylmethionine in pregnant rats]. Nutrición Hospitalaria 1998, 13, 41–49. [Google Scholar]
- Seyoum, G.; Persaud, T. In vitro effect of s-adenosyl methionine on ethanol embryopathy in the rat. Exp. Toxicol. Pathol. 1994, 46, 177–181. [Google Scholar] [CrossRef]
- Yerby, M.S. Management issues for women with epilepsy: Neural tube defects and folic acid supplementation. Neurology 2003, 61, S23–S26. [Google Scholar] [CrossRef]
- Baxter, P. Valproate and folic acid in pregnancy: Associations with autism. Dev. Med. Child Neurol. 2014, 56, 604. [Google Scholar] [CrossRef]
- Turgut, U.; Kazan, S.; Cakin, H.; Ozak, A. Valproic acid effect on neural tube defects is not prevented by concomitant folic acid supplementation: Early chick embryo model pilot study. Int. J. Dev. Neurosci. 2019, 78, 45–48. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Tfilin, M.; Ergaz, Z.; Yanai, J.; Szyf, M.; Turgeman, G. S-adenosyl methionine prevents ASD like behaviors triggered by early postnatal valproic acid exposure in very young mice. Neurotoxicol. Teratol. 2019, 71, 64–74. [Google Scholar] [CrossRef]
- Weinstein-Fudim, L.; Ergaz, Z.; Turgeman, G.; Yanai, J.; Szyf, M.; Ornoy, A. Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior? Int. J. Mol. Sci. 2019, 20, 5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, A.; Howell, K.R.; O Ahmed, A.; Weinberg, D.; Allen, K.M.; Bruggemann, J.; Lenroot, R.; Liu, D.; Galletly, C.; Weickert, C.S.; et al. Association of serum VEGF levels with prefrontal cortex volume in schizophrenia. Mol. Psychiatry 2015, 21, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Lizano, P.L.; Keshavan, M.S.; Tandon, N.; Mathew, I.T.; Mothi, S.S.; Montrose, D.M.; Yao, J.K. Angiogenic and immune signatures in plasma of young relatives at familial high-risk for psychosis and first-episode patients: A preliminary study. Schizophr. Res. 2015, 170, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-C.; Lee, K.-Y.; Kim, Y.-J.; Kim, S.H.; Koh, S.-H.; Lee, Y.J. Serum VEGF levels in acute ischaemic strokes are correlated with long-term prognosis. Eur. J. Neurol. 2009, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, E.R.; Dumitrescu, L.; Moore, A.M.; Cambronero, F.E.; De Jager, P.L.; Koran, M.E.I.; Petyuk, V.A.; Robinson, R.A.S.; Goyal, S.; Schneider, J.A.; et al. Brain expression of the vascular endothelial growth factor gene family in cognitive aging and alzheimer’s disease. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udo, H.; Yoshida, Y.; Kino, T.; Ohnuki, K.; Mizunoya, W.; Mukuda, T.; Sugiyama, H. Enhanced Adult Neurogenesis and Angiogenesis and Altered Affective Behaviors in Mice Overexpressing Vascular Endothelial Growth Factor 120. J. Neurosci. 2008, 28, 14522–14536. [Google Scholar] [CrossRef]
- Weinstein-Fudim, L.; Ergaz, Z.; Szyf, M.; Ornoy, A. Prenatal S-Adenosine Methionine (SAMe) Induces Changes in Gene Expression in the Brain of Newborn Mice That Are Prevented by Co-Administration of Valproic Acid (VPA). Int. J. Mol. Sci. 2020, 21, 2834. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, J.W.; Maruoka, S.; Boon, K.; Garantziotis, S.; Li, Z.; Tomfohr, J.; Bailey, N.; Potts, E.N.; Whitehead, G.; Brass, D.M.; et al. In utero supplementation with methyl donors enhances allergic airway disease in mice. J. Clin. Investig. 2008, 118, 3462–3469. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.L.; Veenstra, G.J.C.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Bahous, R.H.; Jadavji, N.M.; Deng, L.; Cosín-Tomàs, M.; Lu, J.; Malysheva, O.; Leung, K.-Y.; Ho, M.-K.; Pallàs, M.; Kaliman, P.; et al. High dietary folate in pregnant mice leads to pseudo-MTHFR deficiency and altered methyl metabolism, with embryonic growth delay and short-term memory impairment in offspring. Hum. Mol. Genet. 2017, 26, 888–900. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ornoy, A.; Becker, M.; Weinstein-Fudim, L.; Ergaz, Z. S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy. Int. J. Mol. Sci. 2020, 21, 3721. https://doi.org/10.3390/ijms21103721
Ornoy A, Becker M, Weinstein-Fudim L, Ergaz Z. S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy. International Journal of Molecular Sciences. 2020; 21(10):3721. https://doi.org/10.3390/ijms21103721
Chicago/Turabian StyleOrnoy, Asher, Maria Becker, Liza Weinstein-Fudim, and Zivanit Ergaz. 2020. "S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy" International Journal of Molecular Sciences 21, no. 10: 3721. https://doi.org/10.3390/ijms21103721
APA StyleOrnoy, A., Becker, M., Weinstein-Fudim, L., & Ergaz, Z. (2020). S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy. International Journal of Molecular Sciences, 21(10), 3721. https://doi.org/10.3390/ijms21103721