How Do Post-Translational Modifications Influence the Pathomechanistic Landscape of Huntington’s Disease? A Comprehensive Review




Abstract
:1. Introduction to Huntington’s Disease
2. Post-Translational Modifications in Selected Cellular Events of the Diverse Pathomechanism of Huntington’s Disease
2.1. PTMs in Abnormal HTT Protein Aggregation
2.2. Disrupted Proteolytic Pathways: PTMs in Abnormal HTT Protein Degradation
2.3. Tau Impairment and Aberrant Cytoskeleton in HD
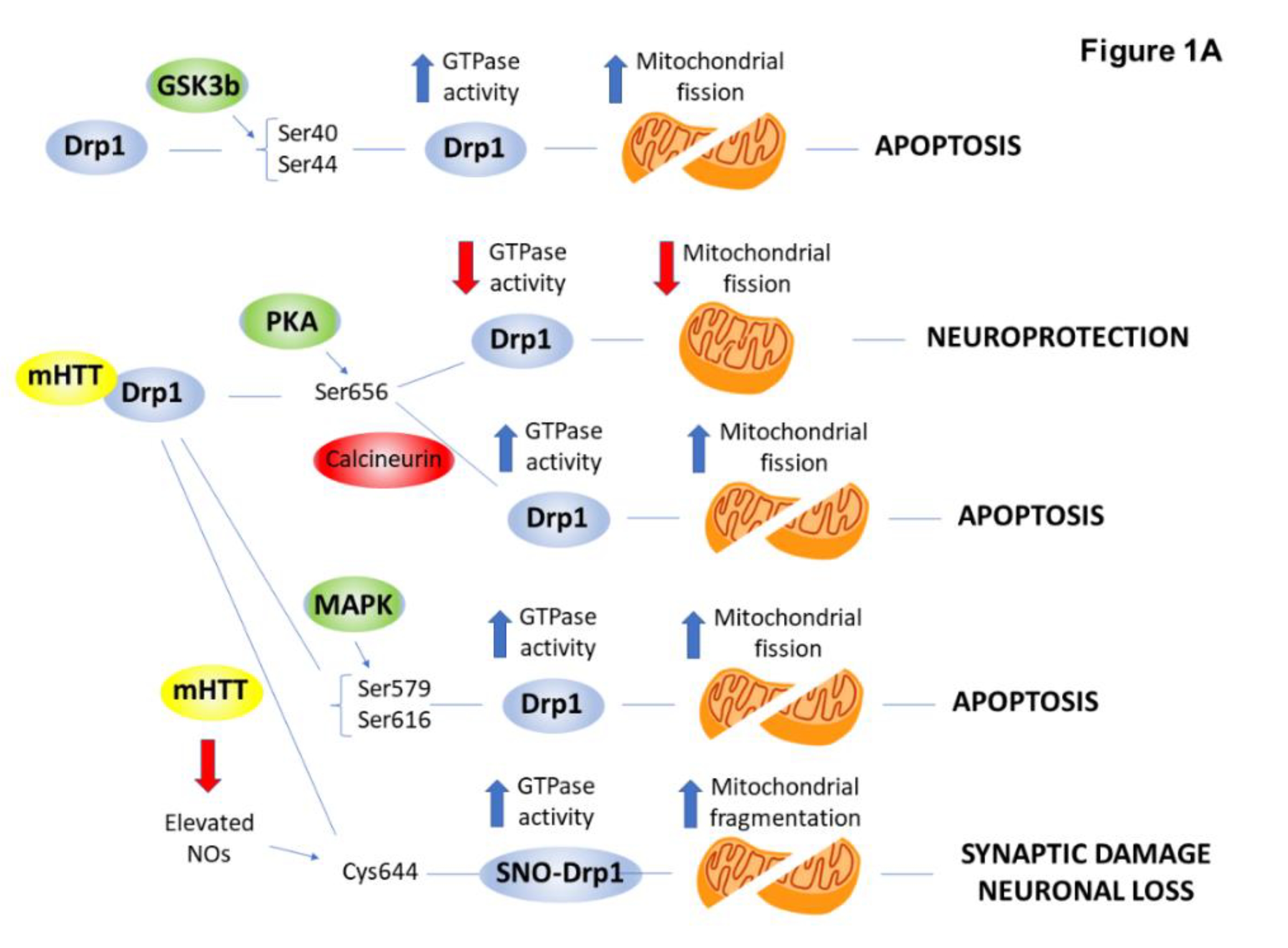
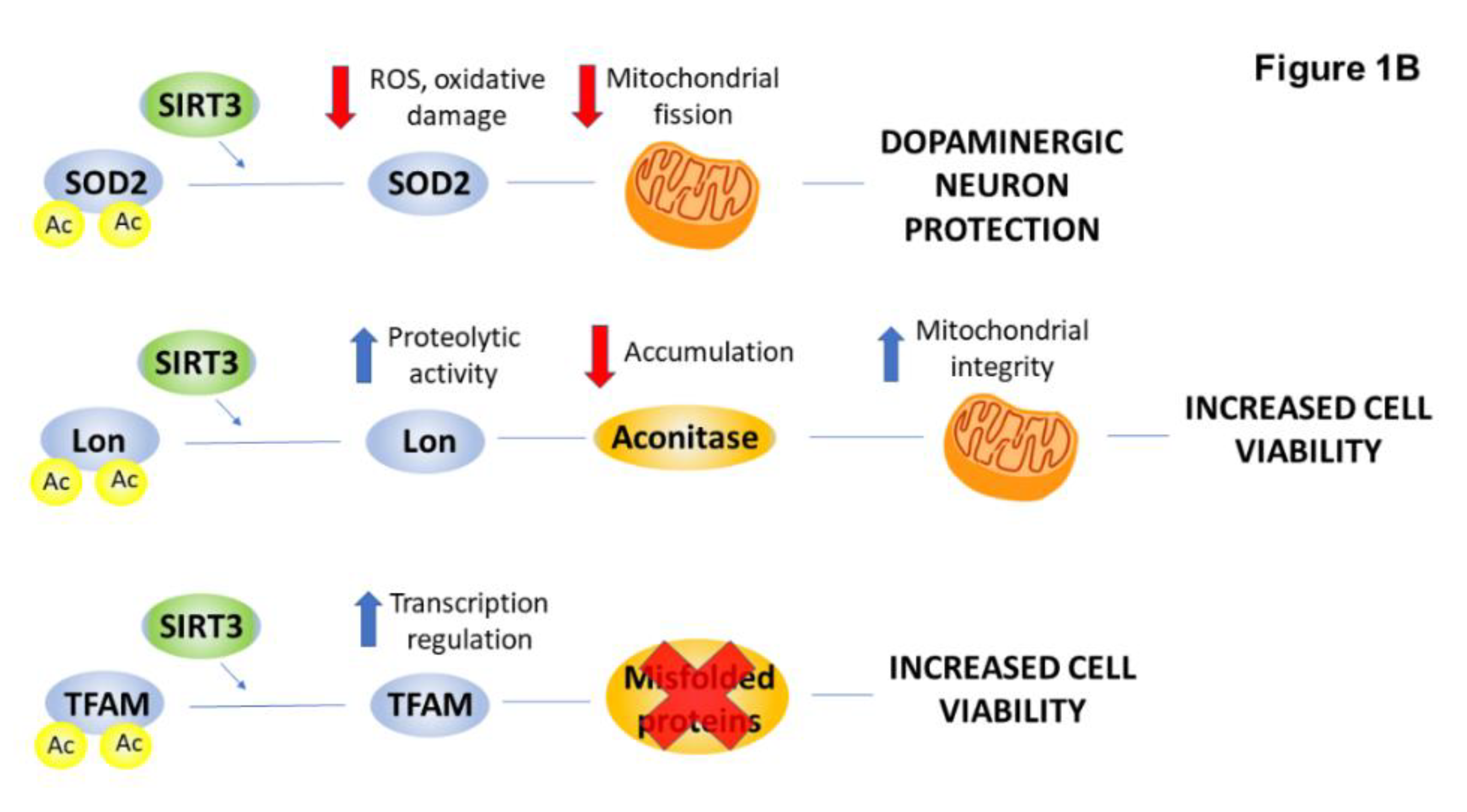
2.4. PTMs Associated with Mitochondrial Abnormalities and Defects in Energy Metabolism in HD
2.5. Cell Death: Focus on Excitotoxicity
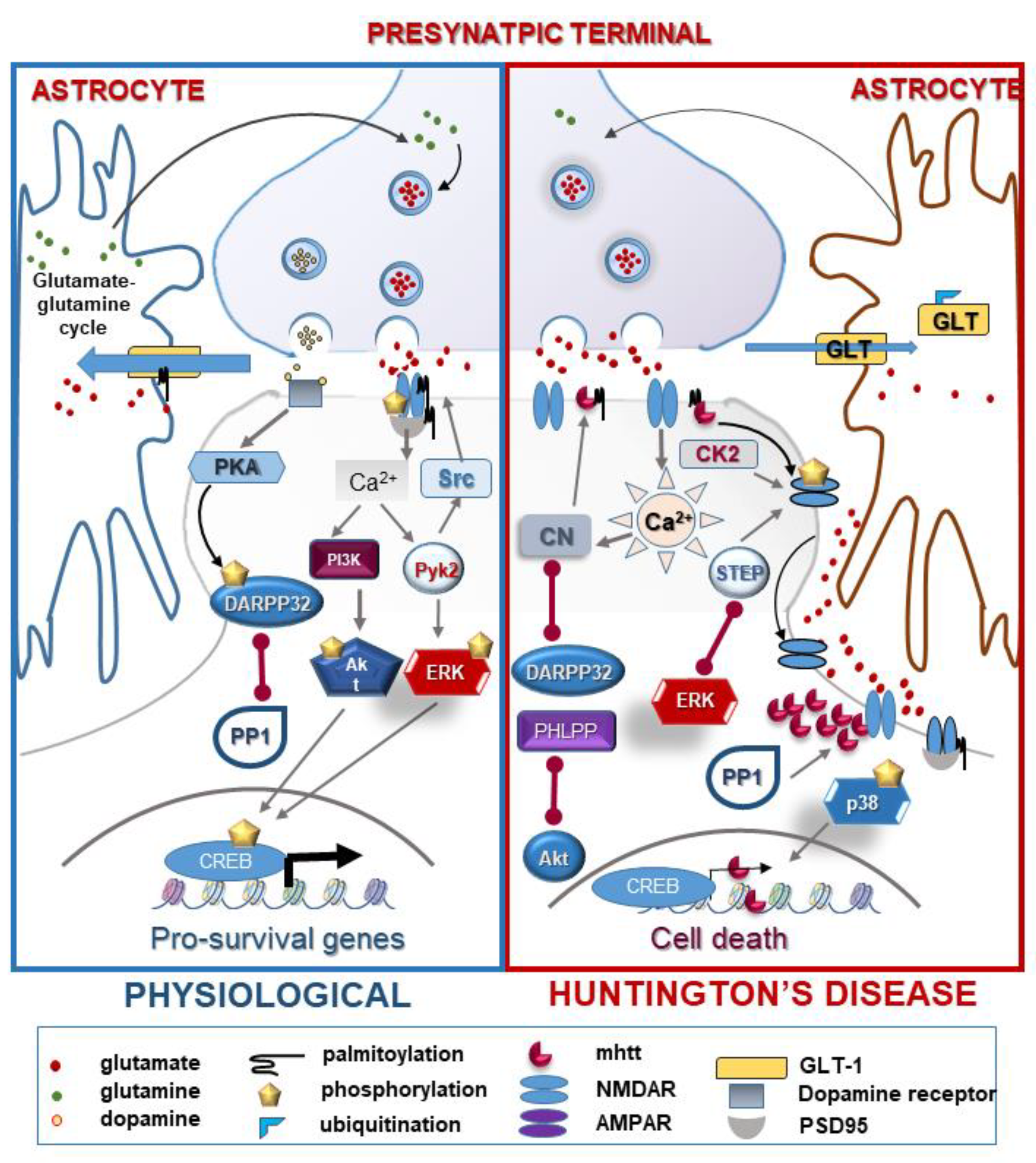
2.5.1. NMDA Receptor: Function and Regulation by PTMs in HD
2.5.2. Ion Channels: AMPA, TRPC5
2.5.3. Glutamate Homeostasis
2.6. Neuroinflammatory Pathways Linked to the Progression of HD
2.7. Transcriptional Dysregulation and Related PTMs in HD
3. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitman, J.; Barak, B.; Benyair, R.; Shenkman, M.; Ashery, U.; Hartl, F.U.; Lederkremer, G.Z. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS ONE 2014, 9, e90803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitman, J.; Ulrich Hartl, F.; Lederkremer, G.Z. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 2013, 4, 2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.M.; Jekabsons, M.B.; Chen, S.; Lin, A.; Rego, A.C.; Gonçalves, J.; Ellerby, L.M.; Nicholls, D.G. Mitochondrial dysfunction in Huntington’s disease: The bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 2007, 101, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Schaffert, L.N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Arbez, N.; Ratovitski, T.; Roby, E.; Chighladze, E.; Stewart, J.C.; Ren, M.; Wang, X.; Lavery, D.J.; Ross, C.A. Post-translational modifications clustering within proteolytic domains decrease mutant huntingtin toxicity. J. Biol. Chem. 2017, 292, 19238–19249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaibva, M.; Jawahery, S.; Pilkington, A.W.; Arndt, J.R.; Sarver, O.; Valentine, S.; Matysiak, S.; Legleiter, J. Acetylation within the First 17 Residues of Huntingtin Exon 1 Alters Aggregation and Lipid Binding. Biophys. J. 2016, 111, 349–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sap, K.A.; Guler, A.T.; Bezstarosti, K.; Bury, A.E.; Juenemann, K.; Demmers, J.A.A.; Reits, E.A. Global Proteome and Ubiquitinome Changes in the Soluble and Insoluble Fractions of Q175 Huntington Mice Brains. Mol. Cell Proteom. 2019, 18, 1705–1720. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.A.; et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.D.O.; Kay, C.; Collins, J.A.; Nguyen, Y.T.; Slama, R.A.; Hayden, M.R. A human huntingtin SNP alters post-translational modification and pathogenic proteolysis of the protein causing Huntington disease. Sci. Rep. 2018, 8, 8096. [Google Scholar] [CrossRef] [Green Version]
- Skotte, N.H.; Sanders, S.S.; Singaraja, R.R.; Ehrnhoefer, D.E.; Vaid, K.; Qiu, X.; Kannan, S.; Verma, C.; Hayden, M.R. Palmitoylation of caspase-6 by HIP14 regulates its activation. Cell Death Differ. 2017, 24, 433–444. [Google Scholar] [CrossRef]
- Martin, D.D.O.; Schmidt, M.E.; Nguyen, Y.T.; Lazic, N.; Hayden, M.R. Identification of a novel caspase cleavage site in huntingtin that regulates mutant huntingtin clearance. FASEB J. 2019, 33, 3190–3197. [Google Scholar] [CrossRef]
- Carbo, M.; Brandi, V.; Pascarella, G.; Staid, D.S.; Colotti, G.; Polticelli, F.; Ilari, A.; Morea, V. Bioinformatics analysis of Ras homologue enriched in the striatum, a potential target for Huntington’s disease therapy. Int. J. Mol. Med. 2019, 44, 2223–2233. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Subramaniam, S. Rhes travels from cell to cell and transports Huntington disease protein via TNT-like protrusion. J. Cell Biol. 2019, 218, 1972–1993. [Google Scholar] [CrossRef] [Green Version]
- Ohta, E.; Itoh, M.; Ueda, M.; Hida, Y.; Wang, M.X.; Hayakawa-Ogura, M.; Li, S.; Nishida, E.; Ohta, K.; Islam, S.; et al. Cullin-4 B E3 ubiquitin ligase mediates Apaf-1 ubiquitination to regulate caspase-9 activity. PLoS ONE 2019, 14, e0219782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.; Ferrer, I.; Rozemuller, A.J.; Hernández, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Jin, N.; Gu, J.; Shi, J.; Zhou, J.; Gong, C.X.; Iqbal, K.; Grundke-Iqbal, I.; Liu, F. Dual-specificity tyrosine phosphorylation-regulated kinase 1 A (Dyrk1A) modulates serine/arginine-rich protein 55 (SRp55)-promoted Tau exon 10 inclusion. J. Biol. Chem. 2012, 287, 30497–30506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratuze, M.; Noël, A.; Julien, C.; Cisbani, G.; Milot-Rousseau, P.; Morin, F.; Dickler, M.; Goupil, C.; Bezeau, F.; Poitras, I.; et al. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Smith, B.R.; Huang, E.S.; Mahesh, A.; Vonsattel, J.P.G.; Petersen, A.J.; Gomez-Pastor, R.; Ashe, K.H. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients. Acta Neuropathol. Commun. 2019, 7, 111. [Google Scholar] [CrossRef]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelières, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; de Proença, J.; Soares, T.R.; Leitão-Rocha, A.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim. Biophys. Acta 2015, 1852, 2484–2493. [Google Scholar] [CrossRef]
- Yue, F.; Li, W.; Zou, J.; Chen, Q.; Xu, G.; Huang, H.; Xu, Z.; Zhang, S.; Gallinari, P.; Wang, F.; et al. Blocking the association of HDAC4 with MAP1 S accelerates autophagy clearance of mutant Huntingtin. Aging (Albany NY) 2015, 7, 839–853. [Google Scholar] [CrossRef] [Green Version]
- Brito, V.; Giralt, A.; Masana, M.; Royes, A.; Espina, M.; Sieiro, E.; Alberch, J.; Castañé, A.; Girault, J.A.; Ginés, S. Cyclin-Dependent Kinase 5 Dysfunction Contributes to Depressive-like Behaviors in Huntington’s Disease by Altering the DARPP-32 Phosphorylation Status in the Nucleus Accumbens. Biol. Psychiatry 2019, 86, 196–207. [Google Scholar] [CrossRef]
- Guo, M.Y.; Shang, L.; Hu, Y.Y.; Jiang, L.P.; Wan, Y.Y.; Zhou, Q.Q.; Zhang, K.; Liao, H.F.; Yi, J.L.; Han, X.J. The role of Cdk5-mediated Drp1 phosphorylation in Abeta1-42 induced mitochondrial fission and neuronal apoptosis. J. Cell Biochem. 2018, 119, 4815–4825. [Google Scholar] [CrossRef]
- Hyun, H.W.; Min, S.J.; Kim, J.E. CDK5 inhibitors prevent astroglial apoptosis and reactive astrogliosis by regulating PKA and DRP1 phosphorylations in the rat hippocampus. Neurosci. Res. 2017, 119, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Roe, A.J.; Qi, X. Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2018, 496, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Haun, F.; Nakamura, T.; Shiu, A.D.; Cho, D.H.; Tsunemi, T.; Holland, E.A.; La Spada, A.R.; Lipton, S.A. S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid. Redox Signal. 2013, 19, 1173–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.; Lipton, S.A. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, H.; Yan, W.Y.; Lei, Y.H.; Wan, Z.; Hou, Y.Y.; Sun, L.K.; Zhou, J.P. SIRT3 Regulation of Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Aging Neurosci. 2019, 11, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neo, S.H.; Tang, B.L. Sirtuins as Modifiers of Huntington’s Disease (HD) Pathology. Prog. Mol. Biol. Transl. Sci. 2018, 154, 105–145. [Google Scholar] [PubMed]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Aerobic glycolysis in amyotrophic lateral sclerosis and Huntington’s disease. Rev. Neurosci. 2018, 29, 547–555. [Google Scholar] [CrossRef]
- Colin, E.; Regulier, E.; Perrin, V.; Durr, A.; Brice, A.; Aebischer, P.; Deglon, N.; Humbert, S.; Saudou, F. Akt is altered in an animal model of Huntington’s disease and in patients. Eur. J. Neurosci. 2005, 21, 1478–1488. [Google Scholar] [CrossRef]
- Saavedra, A.; Garcia-Martinez, J.M.; Xifro, X.; Giralt, A.; Torres-Peraza, J.F.; Canals, J.M.; Diaz-Hernandez, M.; Lucas, J.J.; Alberch, J.; Perez-Navarro, E. PH domain leucine-rich repeat protein phosphatase 1 contributes to maintain the activation of the PI3K/Akt pro-survival pathway in Huntington’s disease striatum. Cell Death Differ. 2010, 17, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.M.; Moser, R.; Régulier, E.; Breuillaud, L.; Dixon, M.; Beesen, A.A.; Elliston, L.; Santos, M.D.F.S.; Kim, J.; Jones, L.; et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J. Neurosci. 2013, 33, 2313–2325. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.M.; Downer, E.J.; Toulouse, A.; Nolan, Y.M. Mitogen-Activated Protein Kinase Phosphatase (MKP)-1 in Nervous System Development and Disease. Mol. Neurobiol. 2015, 51, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Kamceva, M.; Benedict, J.; Nairn, A.C.; Lombroso, P.J. Role of Striatal-Enriched Tyrosine Phosphatase in Neuronal Function. Neural Plast. 2016, 2016, 8136925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissel, B.; Krupp, J.J.; Heinemann, S.F.; Westbrook, G.L. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 2001, 4, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, Y.; Parsons, C.G.; Liu, Y.F. Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-D-aspartate receptors. J. Biol. Chem. 2003, 278, 33364–33369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Wang, L.; Sanders, S.S.; Zuo, K.; Hayden, M.R.; Raymond, L.A. Altered Regulation of Striatal Neuronal N-Methyl-D-Aspartate Receptor Trafficking by Palmitoylation in Huntington Disease Mouse Model. Front. Synaptic Neurosci. 2019, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svenningsson, P.; Nishi, A.; Fisone, G.; Girault, J.A.; Nairn, A.C.; Greengard, P. DARPP-32: An integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 269–296. [Google Scholar] [CrossRef]
- Bibb, J.A.; Yan, Z.; Svenningsson, P.; Snyder, G.L.; Pieribone, V.A.; Horiuchi, A.; Nairn, A.C.; Messer, A.; Greengard, P. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. USA 2000, 97, 6809–6814. [Google Scholar] [CrossRef] [Green Version]
- Parsons, M.P.; Kang, R.; Buren, C.; Dau, A.; Southwell, A.L.; Doty, C.N.; Sanders, S.S.; Hayden, M.R.; Raymond, L.A. Bidirectional control of postsynaptic density-95 (PSD-95) clustering by Huntingtin. J. Biol. Chem. 2014, 289, 3518–3528. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, N.M.; Bordelon, Y.; Brickman, A.M.; Angulo, S.; Khan, U.; Muraskin, J.; Griffith, E.Y.; Wasserman, P.; Menalled, L.; Vonsattel, J.P.; et al. Regional vulnerability in Huntington’s disease: fMRI-guided molecular analysis in patients and a mouse model of disease. Neurobiol. Dis. 2013, 52, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Seo, H.; Kwak, M.; Jeon, J.; Jang, J.; Jeong, E.M.; Myeong, J.; Hwang, Y.J.; Ha, K.; Kang, M.J.; et al. Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 2015, 138, 3030–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.; Choi, S.H.; Kwak, M.; Jeong, B.; Ko, J.; Park, H.J.; Choi, S.; Jun, J.Y.; So, I. TRPC5 channel instability induced by depalmitoylation protects striatal neurons against oxidative stress in Huntington’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118620. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Kang, M.H.; Askew, C.; Kang, R.; Sanders, S.S.; Wan, J.; Davis, N.G.; Hayden, M.R. Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2010, 40, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Raju, K.; Doulias, P.T.; Evans, P.; Krizman, E.N.; Jackson, J.G.; Horyn, O.; Daikhin, Y.; Nissim, I.; Yudkoff, M.; Nissim, I.; et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci. Signal. 2015, 8, ra68. [Google Scholar] [CrossRef] [Green Version]
- Laedermann, C.J.; Cachemaille, M.; Kirschmann, G.; Pertin, M.; Gosselin, R.D.; Chang, I.; Albesa, M.; Towne, C.; Schneider, B.L.; Kellenberger, S.; et al. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Investig. 2013, 123, 3002–3013. [Google Scholar] [CrossRef] [Green Version]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s Disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar]
- Choi, Y.S.; Lee, B.; Cho, H.Y.; Reyes, I.B.; Pu, X.A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington’s disease. Neurobiol. Dis. 2009, 36, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Chakraborty, A.; Geater, C.; Pradhan, S.; Gordon, K.L.; Snowden, J.; Yuan, S.; Dickey, A.S.; Choudhary, S.; Ashizawa, T.; et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife 2019, 8, e42988. [Google Scholar] [CrossRef]
- Lee, M.; Ban, J.J.; Chung, J.Y.; Im, W.; Kim, M. Amelioration of Huntington’s disease phenotypes by Beta-Lapachone is associated with increases in Sirt1 expression, CREB phosphorylation and PGC-1α deacetylation. PLoS ONE 2018, 13, e0195968. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Chandra, A.; Flint Beal, M. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illuzzi, J.L.; Vickers, C.A.; Kmiec, E.B. Modifications of p53 and the DNA damage response in cells expressing mutant form of the protein huntingtin. J. Mol. Neurosci. 2011, 45, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Grison, A.; Mantovani, F.; Comel, A.; Agostoni, E.; Gustincich, S.; Persichetti, F.; Del Sal, G. Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc. Natl. Acad. Sci. USA 2011, 108, 17979–17984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.J.; Han, D.; Kim, K.Y.; Min, S.J.; Kowall, N.W.; Yang, L.; Lee, J.; Kim, Y.; Ryu, H. ESET methylates UBF at K232/254 and regulates nucleolar heterochromatin plasticity and rDNA transcription. Nucleic Acids Res. 2014, 42, 1628–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Hwang, Y.J.; Boo, J.H.; Han, D.; Kwon, O.K.; Todorova, K.; Kowall, N.W.; Kim, Y.; Ryu, H. Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington’s disease. Cell Death Differ. 2011, 18, 1726–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauss, S.; Griesche, N.; Jastrzebska, E.; Chen, C.; Rutschow, D.; Achmuller, C.; Dorn, S.; Boesch, S.M.; Lalowski, M.; Wanker, E.; et al. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat. Commun. 2013, 4, 1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrara, M.; Sigurdardottir, A.; Bertolotti, A. Decoding the selectivity of eIF2 alpha holophosphatases and PPP1 R15A inhibitors. Nat. Struct. Mol. Biol. 2017, 24, 708–716. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Caterino, M.; Squillaro, T.; Montesarchio, D.; Giordano, A.; Giancola, C.; Melone, M.A.B. Huntingtin protein: A new option for fixing the Huntington’s disease countdown clock. Neuropharmacology 2018, 135, 126–138. [Google Scholar] [CrossRef]
- Kim, M.W.; Chelliah, Y.; Kim, S.W.; Otwinowski, Z.; Bezprozvanny, I. Secondary structure of Huntingtin amino-terminal region. Structure 2009, 17, 1205–1212. [Google Scholar] [CrossRef] [Green Version]
- Sivanandam, V.N.; Jayaraman, M.; Hoop, C.L.; Kodali, R.; Wetzel, R.; van der Wel, P.C. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 2011, 133, 4558–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, C.T.; Steffan, J.S.; Guerrero, C.M.; Khashwji, H.; Lukacsovich, T.; Simmons, D.; Purcell, J.M.; Menhaji, K.; Zhu, Y.Z.; Green, K.; et al. Phosphorylation of threonine 3: Implications for Huntingtin aggregation and neurotoxicity. J. Biol. Chem. 2009, 284, 29427–29436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Greiner, E.R.; Mishra, R.; Kodali, R.; Osmand, A.; Finkbeiner, S.; Steffan, J.S.; Thompson, L.M.; Wetzel, R.; Yang, X.W. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 2009, 64, 828–840. [Google Scholar] [CrossRef] [Green Version]
- Cariulo, C.; Azzollini, L.; Verani, M.; Martufi, P.; Boggio, R.; Chiki, A.; Deguire, S.M.; Cherubini, M.; Gines, S.; Marsh, J.L.; et al. Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc. Natl. Acad. Sci. USA 2017, 114, E10809–E10818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiki, A.; DeGuire, S.M.; Ruggeri, F.S.; Sanfelice, D.; Ansaloni, A.; Wang, Z.M.; Cendrowska, U.; Burai, R.; Vieweg, S.; Pastore, A.; et al. Mutant Exon1 Huntingtin Aggregation is Regulated by T3 Phosphorylation-Induced Structural Changes and Crosstalk between T3 Phosphorylation and Acetylation at K6. Angew. Chem. Int. Ed. Engl. 2017, 56, 5202–5207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalinca, H.; Gehin, C.J.C.; Oleinikovas, V.; Lashuel, H.A.; Gervasio, F.L.; Pastore, A. The Role of Post-translational Modifications on the Energy Landscape of Huntingtin N-Terminus. Front. Mol. Biosci. 2019, 6, 95. [Google Scholar] [CrossRef] [Green Version]
- Branco-Santos, J.; Herrera, F.; Poças, G.M.; Pires-Afonso, Y.; Giorgini, F.; Domingos, P.M.; Outeiro, T.F. Protein phosphatase 1 regulates huntingtin exon 1 aggregation and toxicity. Hum. Mol. Genet. 2017, 26, 3763–3775. [Google Scholar] [CrossRef] [Green Version]
- Cariulo, C.; Verani, M.; Martufi, P.; Ingenito, R.; Finotto, M.; Deguire, S.M.; Lavery, D.J.; Toledo-Sherman, L.; Lee, R.; Doherty, E.M.; et al. Ultrasensitive quantitative measurement of huntingtin phosphorylation at residue S13. Biochem. Biophys. Res. Commun. 2020, 521, 549–554. [Google Scholar] [CrossRef]
- DeGuire, S.M.; Ruggeri, F.S.; Fares, M.B.; Chiki, A.; Cendrowska, U.; Dietler, G.; Lashuel, H.A. N-terminal Huntingtin (Htt) phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting. J. Biol. Chem. 2018, 293, 18540–18558. [Google Scholar] [CrossRef] [Green Version]
- Atwal, R.S.; Desmond, C.R.; Caron, N.; Maiuri, T.; Xia, J.; Sipione, S.; Truant, R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 2011, 7, 453–460. [Google Scholar] [CrossRef]
- Ochaba, J.; Fote, G.; Kachemov, M.; Thein, S.; Yeung, S.Y.; Lau, A.L.; Hernandez, S.; Lim, R.G.; Casale, M.; Neel, M.J.; et al. IKKbeta slows Huntington’s disease progression in R6/1 mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10952–10961. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef]
- Bowie, L.E.; Maiuri, T.; Alpaugh, M.; Gabriel, M.; Arbez, N.; Galleguillos, D.; Hung, C.L.K.; Patel, S.; Xia, J.; Hertz, N.T.; et al. N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, E7081–E7090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pardo, A.; Maglione, V.; Alpaugh, M.; Horkey, M.; Atwal, R.S.; Sassone, J.; Ciammola, A.; Steffan, J.S.; Fouad, K.; Truant, R.; et al. Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3528–3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkin, E.E.; Arbez, N.; Waldron-Roby, E.; O’Meally, R.; Ratovitski, T.; Cole, R.N.; Ross, C.A. Phosphorylation of mutant huntingtin at serine 116 modulates neuronal toxicity. PLoS ONE 2014, 9, e88284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Zhang, X.; Liu, H.; LeBron, J.; Alexandris, A.; Peng, Q.; Gu, H.; Yang, F.; Li, Y.; Wang, R.; et al. Nemo-like Kinase Reduces Mutant Huntingtin Levels and Mitigates Huntington’s Disease. Hum. Mol. Genet. 2020. [Google Scholar] [CrossRef]
- Humbert, S.; Bryson, E.A.; Cordelieres, F.P.; Connors, N.C.; Datta, S.R.; Finkbeiner, S.; Greenberg, M.E.; Saudou, F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2002, 2, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Rangone, H.; Poizat, G.; Troncoso, J.; Ross, C.A.; MacDonald, M.E.; Saudou, F.; Humbert, S. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur. J. Neurosci. 2004, 19, 273–279. [Google Scholar] [CrossRef]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [Green Version]
- Zala, D.; Colin, E.; Rangone, H.; Liot, G.; Humbert, S.; Saudou, F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum. Mol. Genet. 2008, 17, 3837–3846. [Google Scholar] [CrossRef] [Green Version]
- Warby, S.C.; Doty, C.N.; Graham, R.K.; Shively, J.; Singaraja, R.R.; Hayden, M.R. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol. Cell Neurosci. 2009, 40, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Gan, L.; Mazarei, G.; Graham, R.K.; Liu, L.; Bissada, N.; Lu, G.; Leavitt, B.R.; Hayden, M.R. Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J. Neurosci. 2010, 30, 14318–14329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, R.; Colin, E.; Regulier, E.; Aebischer, P.; Deglon, N.; Humbert, S.; Saudou, F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J. Neurosci. 2006, 26, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Vacher, C.; Davies, J.E.; Rubinsztein, D.C. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: Implications for mutant huntingtin toxicity. J. Cell Biol. 2005, 169, 647–656. [Google Scholar] [CrossRef]
- Anne, S.L.; Saudou, F.; Humbert, S. Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 2007, 27, 7318–7328. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Forn, M.; Martinez-Torres, S.; Garcia-Diaz Barriga, G.; Alberch, J.; Mila, M.; Azkona, G.; Perez-Navarro, E. Pharmacogenetic modulation of STEP improves motor and cognitive function in a mouse model of Huntington’s disease. Neurobiol. Dis. 2018, 120, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Then, F.; Melia, T.J.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009, 137, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Cong, X.; Held, J.M.; DeGiacomo, F.; Bonner, A.; Chen, J.M.; Schilling, B.; Czerwieniec, G.A.; Gibson, B.W.; Ellerby, L.M. Mass spectrometric identification of novel lysine acetylation sites in huntingtin. Mol. Cell Proteom. 2011, 10, M111.009829. [Google Scholar] [CrossRef] [Green Version]
- Ratovitski, T.; O’Meally, R.N.; Jiang, M.; Chaerkady, R.; Chighladze, E.; Stewart, J.C.; Wang, X.; Arbez, N.; Roby, E.; Alexandris, A.; et al. Post-Translational Modifications (PTMs), Identified on Endogenous Huntingtin, Cluster within Proteolytic Domains between HEAT Repeats. J. Proteome Res. 2017, 16, 2692–2708. [Google Scholar] [CrossRef]
- Varejão, N.; Lascorz, J.; Li, Y.; Reverter, D. Molecular mechanisms in SUMO conjugation. Biochem. Soc. Trans. 2020, 48, 123–135. [Google Scholar] [CrossRef]
- Yau, R.G.; Doerner, K.; Castellanos, E.R.; Haakonsen, D.L.; Werner, A.; Wang, N.; Yang, X.W.; Martinez-Martin, N.; Matsumoto, M.L.; Dixit, V.M.; et al. Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell 2017, 171, 918–933.e20. [Google Scholar] [CrossRef] [Green Version]
- Yau, T.Y.; Molina, O.; Courey, A.J. SUMOylation in development and neurodegeneration. Development 2020, 147. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Vertegaal, A.C. A high-yield double-purification proteomics strategy for the identification of SUMO sites. Nat. Protoc. 2016, 11, 1630–1649. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Princz, A.; Tavernarakis, N. SUMOylation in Neurodegenerative Diseases. Gerontology 2020, 66, 122–130. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.G.; Gareau, J.R.; Ochaba, J.; Song, W.; Raskó, T.; Reverter, D.; Lee, J.; Monteys, A.M.; Pallos, J.; Mee, L.; et al. SUMO-2 and PIAS1 modulate insoluble mutant huntingtin protein accumulation. Cell Rep. 2013, 4, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochaba, J.; Monteys, A.M.; O’Rourke, J.G.; Reidling, J.C.; Steffan, J.S.; Davidson, B.L.; Thompson, L.M. PIAS1 Regulates Mutant Huntingtin Accumulation and Huntington’s Disease-Associated Phenotypes In Vivo. Neuron 2016, 90, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedighi, F.; Adegbuyiro, A.; Legleiter, J. SUMOylation Prevents Huntingtin Fibrillization and Localization onto Lipid Membranes. ACS Chem. Neurosci. 2020, 11, 328–343. [Google Scholar] [CrossRef]
- Huang, K.; Sanders, S.S.; Kang, R.; Carroll, J.B.; Sutton, L.; Wan, J.; Singaraja, R.; Young, F.B.; Liu, L.; El-Husseini, A.; et al. Wild-type HTT modulates the enzymatic activity of the neuronal palmitoyl transferase HIP14. Hum. Mol Genet. 2011, 20, 3356–3365. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Ulrich, H.D.; Sommer, T.; Kaiser, P. The Ubiquitin-Proteasome System of Saccharomyces cerevisiae. Genetics 2012, 192, 319–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Waelter, S.; Boeddrich, A.; Lurz, R.; Scherzinger, E.; Lueder, G.; Lehrach, H.; Wanker, E.E. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 2001, 12, 1393–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.; Sonntag, K.C.; Kim, W.; Cattaneo, E.; Isacson, O. Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS ONE 2007, 2, e238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipper-Krom, S.; Juenemann, K.; Reits, E.A. The Ubiquitin-Proteasome System in Huntington’s Disease: Are Proteasomes Impaired, Initiators of Disease, or Coming to the Rescue? Biochem. Res. Int. 2012, 2012, 837015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juenemann, K.; Jansen, A.H.P.; van Riel, L.; Merkx, R.; Mulder, M.P.C.; An, H.; Statsyuk, A.; Kirstein, J.; Ovaa, H.; Reits, E.A. Dynamic recruitment of ubiquitin to mutant huntingtin inclusion bodies. Sci. Rep. 2018, 8, 1405. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, X.; Gygi, S.P.; Harper, J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 2007, 447, 1135–1138. [Google Scholar] [CrossRef]
- Powis, R.A.; Karyka, E.; Boyd, P.; Côme, J.; Jones, R.A.; Zheng, Y.; Szunyogova, E.; Groen, E.J.; Hunter, G.; Thomson, D.; et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 2016, 1, e87908. [Google Scholar] [CrossRef] [Green Version]
- Wade, B.E.; Wang, C.E.; Yan, S.; Bhat, K.; Huang, B.; Li, S.; Li, X.J. Ubiquitin-activating enzyme activity contributes to differential accumulation of mutant huntingtin in brain and peripheral tissues. J. Neurosci. 2014, 34, 8411–8422. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zeng, L.; Merillat, S.A.; Fischer, S.; Ochaba, J.; Thompson, L.M.; Barmada, S.J.; Scaglione, K.M.; Paulson, H.L. The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington’s disease protein, huntingtin. Neurobiol. Dis. 2018, 109, 127–136. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 353–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Cao, L.; Liang, X.; Du, A.; Peng, T.; Li, H. Herp Promotes Degradation of Mutant Huntingtin: Involvement of the Proteasome and Molecular Chaperones. Mol. Neurobiol. 2018, 55, 7652–7668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Hong, Y.; Yin, P.; Li, S.; Li, X.J. Differential HspBP1 expression accounts for the greater vulnerability of neurons than astrocytes to misfolded proteins. Proc. Natl. Acad. Sci. USA 2017, 114, E7803–E7811. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Jin, Z.; Tan, H.; Xu, Q.; Peng, T.; Li, H. Atypical ubiquitination by E3 ligase WWP1 inhibits the proteasome-mediated degradation of mutant huntingtin. Brain Res. 2016, 1643, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Van Well, E.M.; Bader, V.; Patra, M.; Sánchez-Vicente, A.; Meschede, J.; Furthmann, N.; Schnack, C.; Blusch, A.; Longworth, J.; Petrasch-Parwez, E.; et al. A protein quality control pathway regulated by linear ubiquitination. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Tanji, K.; Mori, F.; Miki, Y.; Utsumi, J.; Sasaki, H.; Kakita, A.; Takahashi, H.; Wakabayashi, K. YOD1 attenuates neurogenic proteotoxicity through its deubiquitinating activity. Neurobiol. Dis. 2018, 112, 14–23. [Google Scholar] [CrossRef]
- Aron, R.; Pellegrini, P.; Green, E.W.; Maddison, D.C.; Opoku-Nsiah, K.; Oliviera, A.O.; Wong, J.S.; Daub, A.C.; Giorgini, F.; Muchowski, P.; et al. Deubiquitinase Usp12 functions noncatalytically to induce autophagy and confer neuroprotection in models of Huntington’s disease. Nat. Commun. 2018, 9, 3191. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 100. [Google Scholar] [CrossRef]
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington’s disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering Mutant Huntingtin Levels and Toxicity: Autophagy-Endolysosome Pathways in Huntington’s Disease. J. Mol. Biol. 2020, 432, 2673–2691. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.M.; Kim, K.; Johnson, C.W.; Chen, S.; Croce, K.R.; Victor, M.B.; Eenjes, E.; Bosco, J.R.; Randolph, L.K.; Dragatsis, I.; et al. Huntington’s Disease Pathogenesis Is Modified In Vivo by Alfy/Wdfy3 and Selective Macroautophagy. Neuron 2020, 105, 813–821.e6. [Google Scholar] [CrossRef]
- Her, L.S.; Lin, J.Y.; Fu, M.H.; Chang, Y.F.; Li, C.L.; Tang, T.Y.; Jhang, Y.L.; Chang, C.Y.; Shih, M.C.; Cheng, P.H.; et al. The Differential Profiling of Ubiquitin-Proteasome and Autophagy Systems in Different Tissues before the Onset of Huntington’s Disease Models. Brain Pathol. 2015, 25, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Kuusisto, E.; Suuronen, T.; Salminen, A. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem. Biophys. Res. Commun. 2001, 280, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Erie, C.; Lu, M.L.; Wei, J. Aberrant subcellular localization of SQSTM1/p62 contributes to increased vulnerability to proteotoxic stress recovery in Huntington’s disease. Mol. Cell Neurosci. 2018, 88, 43–52. [Google Scholar] [CrossRef]
- Kim, E.; Park, S.; Lee, J.H.; Mun, J.Y.; Choi, W.H.; Yun, Y.; Lee, J.; Kim, J.H.; Kang, M.J.; Lee, M.J. Dual Function of USP14 Deubiquitinase in Cellular Proteasomal Activity and Autophagic Flux. Cell Rep. 2018, 24, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Shan, B.; Sun, H.; Xiao, J.; Zhu, K.; Xie, X.; Li, X.; Liang, W.; Lu, X.; Qian, L.; et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016, 30, 1718–1730. [Google Scholar] [CrossRef]
- Srinivasan, V.; Bruelle, C.; Scifo, E.; Pham, D.D.; Soliymani, R.; Lalowski, M.; Lindholm, D. Dynamic Interaction of USP14 with the Chaperone HSC70 Mediates Crosstalk between the Proteasome, ER Signaling, and Autophagy. iScience 2020, 23, 100790. [Google Scholar] [CrossRef] [Green Version]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Trepte, P.; Strempel, N.; Wanker, E.E. Spontaneous self-assembly of pathogenic huntingtin exon 1 protein into amyloid structures. Essays Biochem. 2014, 56, 167–180. [Google Scholar] [PubMed] [Green Version]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Drombosky, K.W.; Rode, S.; Kodali, R.; Jacob, T.C.; Palladino, M.J.; Wetzel, R. Mutational analysis implicates the amyloid fibril as the toxic entity in Huntington’s disease. Neurobiol. Dis. 2018, 120, 126–138. [Google Scholar] [CrossRef]
- Park, K.J.; Grosso, C.A.; Aubert, I.; Kaplan, D.R.; Miller, F.D. p75 NTR-dependent, myelin-mediated axonal degeneration regulates neural connectivity in the adult brain. Nat. Neurosci. 2010, 13, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Herrera, A.E.; Gortat, A.; Carbajo, R.J.; Pineda-Lucena, A.; Orzáez, M.; Pérez-Payá, E. Minocycline inhibits cell death and decreases mutant Huntingtin aggregation by targeting Apaf-1. Hum. Mol. Genet. 2011, 20, 3545–3553. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Kontaxi, C.; Piccardo, P.; Gill, A.C. Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front. Mol. Biosci. 2017, 4, 56. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Maxan, A.; Cicchetti, F. Tau: A Common Denominator and Therapeutic Target for Neurodegenerative Disorders. J. Exp. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Blum, D.; Herrera, F.; Francelle, L.; Mendes, T.; Basquin, M.; Obriot, H.; Demeyer, D.; Sergeant, N.; Gerhardt, E.; Brouillet, E.; et al. Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum. Mol. Genet. 2015, 24, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuono, R.; Winder-Rhodes, S.; de Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A.; REGISTRY Investigators of the European Huntington’s Disease Network. The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain 2015, 138, 1907–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Amour, I.; Turgeon, A.; Goupil, C.; Planel, E.; Hébert, S.S. Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol. 2018, 135, 249–265. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Drouin-Ouellet, J.; Tirolo, C.; Pulvirenti, A.; Giugno, R.; Testa, N.; Caniglia, S.; Serapide, M.F.; Cisbani, G.; Barker, R.A.; et al. GSK-3β-induced Tau pathology drives hippocampal neuronal cell death in Huntington’s disease: Involvement of astrocyte-neuron interactions. Cell Death Dis. 2016, 7, e2206. [Google Scholar] [CrossRef] [Green Version]
- Lim, N.K.; Hung, L.W.; Pang, T.Y.; Mclean, C.A.; Liddell, J.R.; Hilton, J.B.; Li, Q.X.; White, A.R.; Hannan, A.J.; Crouch, P.J. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain. Hum. Mol. Genet. 2014, 23, 4051–4063. [Google Scholar] [CrossRef] [Green Version]
- Elbaz, E.M.; Helmy, H.S.; El-Sahar, A.E.; Saad, M.A.; Sayed, R.H. Lercanidipine boosts the efficacy of mesenchymal stem cell therapy in 3-NP-induced Huntington’s disease model rats via modulation of the calcium/calcineurin/NFATc4 and Wnt/β-catenin signalling pathways. Neurochem. Int. 2019, 131, 104548. [Google Scholar] [CrossRef]
- Muñoz-Lasso, D.C.; Romá-Mateo, C.; Pallardó, F.V.; Gonzalez-Cabo, P. Much More Than a Scaffold: Cytoskeletal Proteins in Neurological Disorders. Cells 2020, 9, 358. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Yoshihara, M.; Littleton, J.T. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3224–3229. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yang, X.J. Tubulin acetylation: Responsible enzymes, biological functions and human diseases. Cell Mol. Life Sci. 2015, 72, 4237–4255. [Google Scholar] [CrossRef]
- Bobrowska, A.; Paganetti, P.; Matthias, P.; Bates, G.P. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2011, 6, e20696. [Google Scholar] [CrossRef]
- Ragot, A.; Pietropaolo, S.; Vincent, J.; Delage, P.; Zhang, H.; Allinquant, B.; Leinekugel, X.; Fischer, A.; Cho, Y.H. Genetic deletion of the Histone Deacetylase 6 exacerbates selected behavioral deficits in the R6/1 mouse model for Huntington’s disease. Brain Behav. 2015, 5, e00361. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Dorostkar, M.M. Dendritic Spine Pathology in Neurodegenerative Diseases. Annu. Rev. Pathol. 2016, 11, 221–250. [Google Scholar] [CrossRef]
- Engmann, O.; Giralt, A.; Gervasi, N.; Marion-Poll, L.; Gasmi, L.; Filhol, O.; Picciotto, M.R.; Gilligan, D.; Greengard, P.; Nairn, A.C.; et al. DARPP-32 interaction with adducin may mediate rapid environmental effects on striatal neurons. Nat. Commun. 2015, 6, 10099. [Google Scholar] [CrossRef] [Green Version]
- Djoussé, L.; Knowlton, B.; Cupples, L.A.; Marder, K.; Shoulson, I.; Myers, R.H. Weight loss in early stage of Huntington’s disease. Neurology 2002, 59, 1325–1330. [Google Scholar] [CrossRef]
- Jenkins, B.G.; Andreassen, O.A.; Dedeoglu, A.; Leavitt, B.; Hayden, M.; Borchelt, D.; Ross, C.A.; Ferrante, R.J.; Beal, M.F. Effects of CAG repeat length, HTT protein length and protein context on cerebral metabolism measured using magnetic resonance spectroscopy in transgenic mouse models of Huntington’s disease. J. Neurochem. 2005, 95, 553–562. [Google Scholar] [CrossRef]
- Dubinsky, J.M. Towards an Understanding of Energy Impairment in Huntington’s Disease Brain. J. Huntingt. Dis. 2017, 6, 267–302. [Google Scholar] [CrossRef] [Green Version]
- Aladdin, A.; Király, R.; Boto, P.; Regdon, Z.; Tar, K. Juvenile Huntington’s Disease Skin Fibroblasts Respond with Elevated Parkin Level and Increased Proteasome Activity as a Potential Mechanism to Counterbalance the Pathological Consequences of Mutant Huntingtin Protein. Int. J. Mol. Sci. 2019, 20, 5338. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.V.; Lund, S.; Greenamyre, J.T. Ca2+-induced permeability transition in human lymphoblastoid cell mitochondria from normal and Huntington’s disease individuals. Mol. Cell. Biochem. 2005, 269, 143–152. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Pinho, B.R.; Soares, T.R.; de Proença, J.; Duchen, M.R.; Oliveira, J.M. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol. Dis. 2016, 90, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef]
- Wang, H.; Lim, P.J.; Karbowski, M.; Monteiro, M.J. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 2009, 18, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Huang, Z.; Xie, F.; Chen, L. Dynamin-related protein 1: A critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J. Cell Physiol. 2019, 234, 10032–10046. [Google Scholar] [CrossRef]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.; Carafoli, E.; Scorrano, L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Deng, H. SIRT3 Overexpression Inhibits Growth of Kidney Tumor Cells and Enhances Mitochondrial Biogenesis. J. Proteome Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 2016, 7, 12646. [Google Scholar] [CrossRef]
- Guidetti, P.; Charles, V.; Chen, E.Y.; Reddy, P.H.; Kordower, J.H.; Whetsell, W.O., Jr.; Schwarcz, R.; Tagle, D.A. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp. Neurol. 2001, 169, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N. Mutant Huntingtin and Elusive Defects in Oxidative Metabolism and Mitochondrial Calcium Handling. Mol. Neurobiol. 2016, 53, 2944–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, A.A.; Lee, D.Y.; Datta, R.; Hauser, M.; Budworth, H.; Holt, A.; Mihalik, S.; Goldschmidt, P.; Frankel, K.; Trego, K.; et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab. 2019, 29, 1258–1273.e11. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Schapira, A.H. Secondary abnormalities of mitochondrial DNA associated with neurodegeneration. Biochem. Soc. Symp. 1999, 66, 99–110. [Google Scholar]
- Lou, S.; Lepak, V.C.; Eberly, L.E.; Roth, B.; Cui, W.; Zhu, X.H.; Oz, G.; Dubinsky, J.M. Oxygen consumption deficit in Huntington disease mouse brain under metabolic stress. Hum. Mol. Genet. 2016, 25, 2813–2826. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. New techniques for investigating mitochondrial DNA in neurodegenerative diseases. Neurology 1997, 49, 907–908. [Google Scholar] [CrossRef]
- Manfredi, G.; Beal, M.F. The role of mitochondria in the pathogenesis of neurodegenerative diseases. Brain Pathol. 2000, 10, 462–472. [Google Scholar] [CrossRef]
- Jenkins, B.G.; Koroshetz, W.J.; Beal, M.F.; Rosen, B.R. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1 H NMR spectroscopy. Neurology 1993, 43, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Bryan, M.R.; Nordham, K.D.; Rose, D.I.R.; O’Brien, M.T.; Joshi, P.; Foshage, A.M.; Goncalves, F.M.; Nitin, R.; Uhouse, M.A.; Aschner, M.; et al. Manganese Acts upon Insulin/IGF Receptors to Phosphorylate AKT and Increase Glucose Uptake in Huntington’s Disease Cells. Mol. Neurobiol. 2020, 57, 1570–1593. [Google Scholar] [CrossRef]
- Naia, L.; Ferreira, I.L.; Cunha-Oliveira, T.; Duarte, A.I.; Ribeiro, M.; Rosenstock, T.R.; Laco, M.N.; Ribeiro, M.J.; Oliveira, C.R.; Saudou, F.; et al. Activation of IGF-1 and insulin signaling pathways ameliorate mitochondrial function and energy metabolism in Huntington’s Disease human lymphoblasts. Mol. Neurobiol. 2015, 51, 331–348. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kowall, N.W.; Beal, M.F.; Martin, J.B.; Bird, E.D.; Richardson, E.P., Jr. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1987, 46, 12–27. [Google Scholar] [CrossRef]
- Fan, M.M.; Raymond, L.A. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog. Neurobiol. 2007, 81, 272–293. [Google Scholar] [CrossRef]
- Schwarcz, R.; Coyle, J.T. Striatal lesions with kainic acid: Neurochemical characteristics. Brain Res. 1977, 127, 235–249. [Google Scholar] [CrossRef]
- Freese, A.; DiFiglia, M.; Koroshetz, W.J.; Beal, M.F.; Martin, J.B. Characterization and mechanism of glutamate neurotoxicity in primary striatal cultures. Brain Res. 1990, 521, 254–264. [Google Scholar] [CrossRef]
- Gardian, G.; Vecsei, L. Huntington’s disease: Pathomechanism and therapeutic perspectives. J. Neural Transm. (Vienna) 2004, 111, 1485–1494. [Google Scholar] [CrossRef]
- Johnson, J.W.; Ascher, P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–531. [Google Scholar] [CrossRef]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [Google Scholar] [CrossRef]
- MacDermott, A.B.; Mayer, M.L.; Westbrook, G.L.; Smith, S.J.; Barker, J.L. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 1986, 321, 519–522. [Google Scholar] [CrossRef]
- Koutsilieri, E.; Riederer, P. Excitotoxicity and new antiglutamatergic strategies in Parkinson’s disease and Alzheimer’s disease. Parkinsonism Relat. Disord. 2007, 13 (Suppl. 3), S329–S331. [Google Scholar] [CrossRef]
- Berliocchi, L.; Fava, E.; Leist, M.; Horvat, V.; Dinsdale, D.; Read, D.; Nicotera, P. Botulinum neurotoxin C initiates two different programs for neurite degeneration and neuronal apoptosis. J. Cell Biol. 2005, 168, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, C.; Virág, L. Inputs and outputs of poly(ADP-ribosyl)ation: Relevance to oxidative stress. Redox Biol. 2014, 2, 978–982. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, T.; Mocle, A.J.; Hung, C.L.; Xia, J.; van Roon-Mom, W.M.; Truant, R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum. Mol. Genet. 2017, 26, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Virág, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef]
- Vis, J.C.; Schipper, E.; de Boer-van Huizen, R.T.; Verbeek, M.M.; de Waal, R.M.; Wesseling, P.; ten Donkelaar, H.J.; Kremer, B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol. 2005, 109, 321–328. [Google Scholar] [CrossRef]
- Cardinale, A.; Paldino, E.; Giampa, C.; Bernardi, G.; Fusco, F.R. PARP-1 Inhibition Is Neuroprotective in the R6/2 Mouse Model of Huntington’s Disease. PLoS ONE 2015, 10, e0134482. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Mandir, A.S.; Poitras, M.F.; Berliner, A.R.; Herring, W.J.; Guastella, D.B.; Feldman, A.; Poirier, G.G.; Wang, Z.Q.; Dawson, T.M.; Dawson, V.L. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J. Neurosci. 2000, 20, 8005–8011. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Vijayan, R.; Sekar, S.; Mani, S.; Rajamani, B.; Ganapathy, R. Simultaneous blockade of NMDA receptors and PARP-1 activity synergistically alleviate immunoexcitotoxicity and bioenergetics in 3-nitropropionic acid intoxicated mice: Evidences from memantine and 3-aminobenzamide interventions. Eur. J. Pharmacol. 2017, 803, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fang, Y. New insights of poly(ADP-ribosylation) in neurodegenerative diseases: A focus on protein phase separation and pathologic aggregation. Biochem. Pharmacol. 2019, 167, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Clemente, A.; Matta, J.A.; Isaac, J.T.; Roche, K.W. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 2010, 67, 984–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, M.W.; Kalia, L.V. Src kinases: A hub for NMDA receptor regulation. Nat. Rev. Neurosci. 2004, 5, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Castilho, R.F.; Korhonen, L.; Lindholm, D.; Bates, G.P.; Brundin, P. Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington’s disease is dependent on age and CAG repeat length. J. Neurochem. 2001, 78, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Giralt, A.; Brito, V.; Chevy, Q.; Simonnet, C.; Otsu, Y.; Cifuentes-Diaz, C.; de Pins, B.; Coura, R.; Alberch, J.; Gines, S.; et al. Pyk2 modulates hippocampal excitatory synapses and contributes to cognitive deficits in a Huntington’s disease model. Nat. Commun. 2017, 8, 15592. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Gray, J.A.; Ogilvie, K.A.; Nicoll, R.A.; Roche, K.W. Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep. 2013, 3, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Tingley, W.G.; Ehlers, M.D.; Kameyama, K.; Doherty, C.; Ptak, J.B.; Riley, C.T.; Huganir, R.L. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem. 1997, 272, 5157–5166. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.Y.; Skeberdis, V.A.; Jover, T.; Grooms, S.Y.; Lin, Y.; Araneda, R.C.; Zheng, X.; Bennett, M.V.; Zukin, R.S. Protein kinase C modulates NMDA receptor trafficking and gating. Nat. Neurosci. 2001, 4, 382–390. [Google Scholar] [CrossRef]
- Scott, D.B.; Blanpied, T.A.; Ehlers, M.D. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 2003, 45, 755–767. [Google Scholar] [CrossRef]
- Ariano, M.A.; Wagle, N.; Grissell, A.E. Neuronal vulnerability in mouse models of Huntington’s disease: Membrane channel protein changes. J. Neurosci. Res. 2005, 80, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Gray, J.A.; Sanz-Clemente, A.; Wei, Z.; Thomas, E.V.; Nicoll, R.A.; Roche, K.W. SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep. 2012, 2, 1120–1128. [Google Scholar] [CrossRef] [Green Version]
- Rusnak, F.; Mertz, P. Calcineurin: Form and function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef] [PubMed]
- Xifro, X.; Garcia-Martinez, J.M.; Del Toro, D.; Alberch, J.; Perez-Navarro, E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J. Neurochem. 2008, 105, 1596–1612. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Ceyzeriat, K.; Carrillo-de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef]
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175. [Google Scholar] [CrossRef]
- Koster, K.P.; Francesconi, W.; Berton, F.; Alahmadi, S.; Srinivas, R.; Yoshii, A. Developmental NMDA receptor dysregulation in the infantile neuronal ceroid lipofuscinosis mouse model. Elife 2019, 8, e40316. [Google Scholar] [CrossRef]
- Yger, M.; Girault, J.A. DARPP-32, Jack of All Trades... Master of Which? Front. Behav. Neurosci. 2011, 5, 56. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, J.D.; Huganir, R.L. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007, 23, 613–643. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, C.; Vincent, J.; Zala, D.; Benstaali, C.; Sainlos, M.; Grillo-Bosch, D.; Daburon, S.; Coussen, F.; Cho, Y.; et al. Modulation of AMPA receptor surface diffusion restores hippocampal plasticity and memory in Huntington’s disease models. Nat. Commun. 2018, 9, 4272. [Google Scholar] [CrossRef]
- Zheng, W.; Yang, J.; Beauchamp, E.; Cai, R.; Hussein, S.; Hofmann, L.; Li, Q.; Flockerzi, V.; Berthiaume, L.G.; Tang, J.; et al. Regulation of TRPP3 Channel Function by N-terminal Domain Palmitoylation and Phosphorylation. J. Biol. Chem. 2016, 291, 25678–25691. [Google Scholar] [CrossRef] [Green Version]
- Levy, L.M.; Lehre, K.P.; Walaas, S.I.; Storm-Mathisen, J.; Danbolt, N.C. Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur. J. Neurosci. 1995, 7, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Sanchez, A.M.; Montiel, T.; Segovia, J.; Massieu, L. Glutamate toxicity in the striatum of the R6/2 Huntington’s disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiol. Dis. 2009, 34, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Silvestroni, A.; Faull, R.L.; Strand, A.D.; Moller, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat. Med. 2006, 12, 1005–1015. [Google Scholar] [PubMed]
- Przedborski, S. Neuroinflammation and Parkinson’s disease. Handb. Clin. Neurol. 2007, 83, 535–551. [Google Scholar] [PubMed]
- Wild, E.; Bjorkqvist, M.; Tabrizi, S.J. Immune markers for Huntington’s disease? Expert Rev. Neurother. 2008, 8, 1779–1781. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef]
- Khoshnan, A.; Patterson, P.H. The role of IkappaB kinase complex in the neurobiology of Huntington’s disease. Neurobiol. Dis. 2011, 43, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Witt, J.; Barisic, S.; Schumann, E.; Allgower, F.; Sawodny, O.; Sauter, T.; Kulms, D. Mechanism of PP2A-mediated IKK beta dephosphorylation: A systems biological approach. BMC Syst. Biol. 2009, 3, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trager, U.; Andre, R.; Lahiri, N.; Magnusson-Lind, A.; Weiss, A.; Grueninger, S.; McKinnon, C.; Sirinathsinghji, E.; Kahlon, S.; Pfister, E.L.; et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain 2014, 137, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Boje, K.M. Nitric oxide neurotoxicity in neurodegenerative diseases. Front. Biosci. 2004, 9, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Yoo, H.S.; Ju, Y.J.; Oh, M.S.; Lee, K.T.; Inn, K.S.; Kim, N.J.; Lee, J.K. Synthetic 3’,4’-Dihydroxyflavone Exerts Anti-Neuroinflammatory Effects in BV2 Microglia and a Mouse Model. Biomol. Ther. (Seoul) 2018, 26, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Trager, U.; Magnusson, A.; Lahiri Swales, N.; Wild, E.; North, J.; Lowdell, M.; Bjorkqvist, M. JAK/STAT Signalling in Huntington’s Disease Immune Cells. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Babcock, A.A.; Wirenfeldt, M.; Holm, T.; Nielsen, H.H.; Dissing-Olesen, L.; Toft-Hansen, H.; Millward, J.M.; Landmann, R.; Rivest, S.; Finsen, B.; et al. Toll-like receptor 2 signaling in response to brain injury: An innate bridge to neuroinflammation. J. Neurosci. 2006, 26, 12826–12837. [Google Scholar] [CrossRef] [Green Version]
- Griffioen, K.; Mattson, M.P.; Okun, E. Deficiency of Toll-like receptors 2, 3 or 4 extends life expectancy in Huntington’s disease mice. Heliyon 2018, 4, e00508. [Google Scholar] [CrossRef] [Green Version]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38 alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta). J. Neuroinflamm. 2011, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Valor, L.M. Transcription, epigenetics and ameliorative strategies in Huntington’s Disease: A genome-wide perspective. Mol. Neurobiol. 2015, 51, 406–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, C.; Zhang, S.; Dong, X.; Ma, S.; Cong, S. Transcriptional Dysregulation and Post-translational Modifications in Polyglutamine Diseases: From Pathogenesis to Potential Therapeutic Strategies. Front. Mol. Neurosci. 2018, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Valor, L.M.; Guiretti, D.; Lopez-Atalaya, J.P.; Barco, A. Genomic landscape of transcriptional and epigenetic dysregulation in early onset polyglutamine disease. J. Neurosci. 2013, 33, 10471–10482. [Google Scholar] [CrossRef] [PubMed]
- Cong, S.Y.; Pepers, B.A.; Evert, B.O.; Rubinsztein, D.C.; Roos, R.A.; van Ommen, G.J.; Dorsman, J.C. Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol. Cell Neurosci. 2005, 30, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Poirier, M.A.; Liang, Y.; Pei, Z.; Weiskittel, C.E.; Smith, W.W.; DeFranco, D.B.; Ross, C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 2006, 23, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Puigdellívol, M.; Carretón, O.; Paoletti, P.; Valero, J.; Parra-Damas, A.; Saura, C.A.; Alberch, J.; Ginés, S. Long-term memory deficits in Huntington’s disease are associated with reduced CBP histone acetylase activity. Hum. Mol. Genet. 2012, 21, 1203–1216. [Google Scholar] [CrossRef] [Green Version]
- Bertogliat, M.J.; Morris-Blanco, K.C.; Vemuganti, R. Epigenetic mechanisms of neurodegenerative diseases and acute brain injury. Neurochem. Int. 2020, 133, 104642. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Zsindely, N.; Faragó, A.; Marsh, J.L.; Bodai, L. Systematic genetic interaction studies identify histone demethylase Utx as potential target for ameliorating Huntington’s disease. Hum. Mol. Genet. 2018, 27, 759. [Google Scholar] [CrossRef] [Green Version]
- Ratovitski, T.; Arbez, N.; Stewart, J.C.; Chighladze, E.; Ross, C.A. PRMT5- mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington’s disease (HD). Cell Cycle 2015, 14, 1716–1729. [Google Scholar] [CrossRef] [Green Version]
- Ben Yehuda, A.; Risheq, M.; Novoplansky, O.; Bersuker, K.; Kopito, R.R.; Goldberg, M.; Brandeis, M. Ubiquitin Accumulation on Disease Associated Protein Aggregates Is Correlated with Nuclear Ubiquitin Depletion, Histone De-Ubiquitination and Impaired DNA Damage Response. PLoS ONE 2017, 12, e0169054. [Google Scholar] [CrossRef] [PubMed]
- Chrivia, J.C.; Kwok, R.P.; Lamb, N.; Hagiwara, M.; Montminy, M.R.; Goodman, R.H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993, 365, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Paldino, E.; Giampà, C.; Montagna, E.; Angeloni, C.; Fusco, F.R. Modulation of Phospho-CREB by Systemically Administered Recombinant BDNF in the Hippocampus of the R6/2 Mouse Model of Huntington’s Disease. Neurosci. J. 2019, 2019, 8363274. [Google Scholar] [CrossRef] [Green Version]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, O.; Chen, C.; Bingham, R.; Argyrou, A.; Buxton, R.; Pancevac Jonsson, C.; Jones, E.; Bridges, A.; Gatfield, K.; Krauss, S.; et al. Pharmacological disruption of the MID1/alpha4 interaction reduces mutant Huntingtin levels in primary neuronal cultures. Neurosci. Lett. 2018, 673, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Rayner, S.L.; Morsch, M.; Molloy, M.P.; Shi, B.; Chung, R.; Lee, A. Using proteomics to identify ubiquitin ligase-substrate pairs: How novel methods may unveil therapeutic targets for neurodegenerative diseases. Cell Mol. Life Sci. 2019, 76, 2499–2510. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target Protein in HD (Abbreviation) | Modification (Enzymes) | Alteration in HD | Affected Cellular Process | Ref. |
|---|---|---|---|---|
| Protein aggregation | ||||
| Huntingtin (HTT) | phosphorylation (IKK,CK2,NLK,Akt, SGK,CDK5/PP1,PP2A, PP2B) | ↓ | mHTT aggregation | [11] |
| acetylation (CBP/HDAC1) | ↓ | formation of fibrillary aggregates, lipid-binding | [11,12] | |
| ubiquitination | ↑/↓ | proteosomal degradation | [13] | |
| SUMOylation (PIAS1, RHES) | ↑ | escape insoluble aggregate formation, neurotoxicity | [14] | |
| palmitoylation | ↓ | inclusion formation | [15] | |
| myristoylation | ↓ | pathogenic proteolysis | [16] | |
| caspase cleavage (caspase-1, -6) | ↑ | mHTT aggregation | [17,18] | |
| Ras Homolog Enriched in Striatum (RHES) | farnesylation | ↑ | abolished SUMOylation of mHTT | [19,20] |
| Proteolytic cleavage | ||||
| Caspase-6 (CASP6) | palmitoylation | ↑ | CASP6 activation | [17] |
| Apoptotic protease-activating factor 1 (Apaf-1) | ubiquitination | ↑ | regulation of caspase-9 | [21] |
| Tau impairment and cytoskeletal alterations | ||||
| Serine/arginine-rich splicing factor-6 (SRSF6, aka SRp55) | phosphorylation (Dyrk1A) | ↑ | faulty splicing of tau | [22,23] |
| Tau | phosphorylation (CDK5/PP2B) | ↑ | tau aggregation | [24] |
| caspase cleavage (caspase-2) | ↑ | tau truncation | [25] | |
| Tubulin | acetylation | ↓ | vesicular transport deficit | [26,27] |
| Microtubule-associated protein 1 S (MAPS1) | acetylation | ↓ | mHTT degradation | [28] |
| β-adducin | phosphorylation (PKA) | ↑ | dendritic spine destabilization | [29] |
| Mitochondrial abnormalities and defects in energy metabolism | ||||
| Dynamin-related protein (Drp1) | phosphorylation (GSK-3β, MAPK1, CDK5/PP2B) | ↑ | mitochondrial fragmentation | [30,31,32] |
| S-nitrosylation | ↑ | mitochondrial fragmentation | [33,34] | |
| Manganese superoxide dismutase (MnSOD) | acetylation | ↓ | mitochondrial biogenesis | [35,36] |
| Lon protease | acetylation | ↓ | degradation of aconitase | [37] |
| β-catenin | phosphorylation (GSK-3, CK1) | ↓ | less efficient energy production | [38] |
| ubiquitination | ↓ | [38] | ||
| Neuroinflammatory pathways | ||||
| Akt | phosphorylation (PI3K/PHLPP2,PP2A) | ↓ | activation of apoptotic signaling pathways | [39,40] |
| JNK/p38 | phosphorylation (MKP-1/DUSP1) | ↑ | loss of neuroprotection | [41,42] |
| Excitotoxicity | ||||
| N-methyl D-aspartate receptors (NMDARs) | phosphorylation | ↑/↓ | excitotoxicity disorder of NMDAR trafficking and ER transport | [43,44,45] |
| palmitoylation | ↓ | increased extrasynaptical localization and cellular death | [46] | |
| Postsynaptic density 95 kDa (PSD-95) | palmitoylation | ↓ | disorder in neuronal development, faulty localization of PSD-95 | [47,48,49] |
| Dopamine- and cAMP-regulated phosphoprotein 32 (DARPP32) | phosphorylation (PKA/PP1,PP2A) | ↓ | enhanced NMDA-induced excitotoxicity | [48,50] |
| TRCP5 | S-palmitoylation | ↑ | excess Ca2+ influx | [51] |
| gluthationylation | ↑ | [52] | ||
| Glutamate transporter-1 (GLT-1) | palmitoylation | ↓ | defect in glutamate uptake | [53] |
| ubiquitination | ↑ | [54] | ||
| nitrosylation | ↑ | [55] | ||
| Transcriptional dysregulation | ||||
| Histones | acetylation | ↓ | altered gene expression | [56] |
| Lys methylation | ↓/↑ | |||
| Arg methylation | ↓ | |||
| ubiquitination | ↓ | |||
| cAMP response element-binding protein (CREB) | phosphorylation | ↓ | repressed BDNF expression | [57,58] |
| CREB binding protein (CBP) | ubiquitination | ↑ | CBP degradation, histone hypoacetylation | [59] |
| Peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) | acetylation | ↑ | mitochondrial dysfunction | [60,61] |
| p53 protein | phosphorylation | ↑ | upregulation of apoptosis-related genes | [62,63] |
| acetylation | ↓ | |||
| Upstream binding factor (UBF) | trimethylation | ↑ | repressed rDNA transcription | [64,65] |
| acetylation | ↓ | |||
| Ribosomal S6 kinase (S6 K) | phosphorylation | ↑ | HTT transcription | [66] |
| Eukaryotic translation initiation factor 2 (eIF2 a) | phosphorylation (PERK/R15A-PP1, R15B-PP1) | protein quality control | [67] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lontay, B.; Kiss, A.; Virág, L.; Tar, K. How Do Post-Translational Modifications Influence the Pathomechanistic Landscape of Huntington’s Disease? A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 4282. https://doi.org/10.3390/ijms21124282
Lontay B, Kiss A, Virág L, Tar K. How Do Post-Translational Modifications Influence the Pathomechanistic Landscape of Huntington’s Disease? A Comprehensive Review. International Journal of Molecular Sciences. 2020; 21(12):4282. https://doi.org/10.3390/ijms21124282
Chicago/Turabian StyleLontay, Beata, Andrea Kiss, László Virág, and Krisztina Tar. 2020. "How Do Post-Translational Modifications Influence the Pathomechanistic Landscape of Huntington’s Disease? A Comprehensive Review" International Journal of Molecular Sciences 21, no. 12: 4282. https://doi.org/10.3390/ijms21124282
APA StyleLontay, B., Kiss, A., Virág, L., & Tar, K. (2020). How Do Post-Translational Modifications Influence the Pathomechanistic Landscape of Huntington’s Disease? A Comprehensive Review. International Journal of Molecular Sciences, 21(12), 4282. https://doi.org/10.3390/ijms21124282