The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome



Abstract
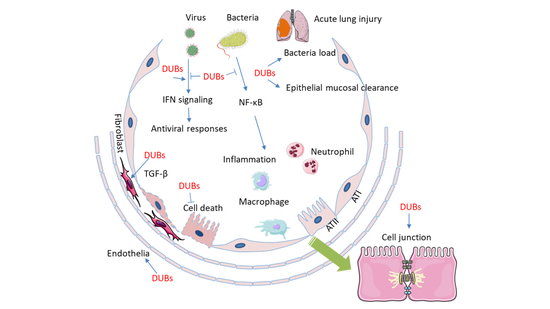
:
1. Introduction
2. Protein Ubiquitin Proteasomal Degradation and Deubiquitination
3. Molecular Mechanisms of DUBs in the Pathogenesis of ALI/ARDS
4. Deubiquitinating Enzymes Involved in ALI/ARDS
4.1. USPs
4.2. OTUs
4.3. JAMMs
4.4. OTHER DUBs
5. Potential Therapeutic Approaches Targeting DUBS in ALI/ARDS
6. Conclusions and Future Perspectives
Funding
Conflicts of Interest
Abbreviations
| ABRO1 | Abraxas Brother 1 |
| ALI | Acute lung injury |
| ALKBH3 | AlkB homologue 3 |
| AMSH | Anti-Müllerian hormone |
| ARDS | Acute respiratory distress syndrome |
| ASC | Apeck-like protein containing a CARD |
| ATXN3 | Ataxin3 |
| BRISC | BRCC36 isopeptidase complex |
| CBP | CREB-binding protein |
| cIAP-1 | Cellular inhibitor of apoptosis protein-1 |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CSN5 | COP9 signalosome 5 |
| COVID-19 | Coronavirus disease 2019 |
| CYLD | Cylindromatosis |
| DUBs | Deubiquitinating enzymes |
| E2F1 | E2F transcription factor 1 |
| EIF3 | Eukaryotic translation initiation factor 3 |
| HBO1 | Histone acetyltransferase binding to origin recognition complex 1 |
| HDAC2 | Histone deacetylase 2 |
| IFN | Interferon |
| IKKγ | IκB kinase γ |
| IL-1β | Interlukin-1β |
| IRF3 | Interferon regulatory factor 3 |
| JAMMs | Zn-JAB1/MPN/MOV34 domain metallopeptidase |
| LPA1 | Lysophosphatidic acid receptor 1 |
| LPS | Lipopolysaccharide |
| LUBAC | Linear ubiquitin chain assembly complex |
| MAVS | Mitochondria antiviral-signaling protein |
| MCL1 | Myeloid cell leukemia 1 |
| MINDYs | Motif interacting with ubiquitin - containing novel DUB family |
| MJDs | Machado-Josephin disease protein domain protease |
| NEMO | Nuclear factor (NF)-κB essential modulator |
| NALP7 | NACHT, LRR and PYD domains-containing protein 7 |
| NFAT | Nuclear factor of activated T cells |
| NICD1 | NOTCH1 intracellular domain |
| NLRP3 | NLR family pyrin domain containing 3 |
| MYSM1 | MPN domain containing (MPND, myb-like SWIRM and MPN domains 1 |
| OTUs | Ovarian tumor proteases |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PEG10 | Paternally expressed gene 10 |
| PLY | Pneumolysin |
| POH1 | Proteasome non-ATPase regulatory subunit 14 |
| PRPF8 | Pre-mRNA-processing-splicing factor 8 |
| PSMD7 | Proteasome non-ATPase regulatory subunit 7 |
| RIG-1 | Retinoic acid-inducible gene I |
| RIPK1 | Receptor-interacting protein kinase 1 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| STAMBP | STAM-binding protein |
| STING | Stimulator of interferon |
| TAK1 | TGF-β-activated kinase 1 |
| TGFβ-1 | Transforming growth factor β-1 |
| TRIF | TIR domain-containing adaptor inducing interferon-β |
| TNF-α | Tumor necrosis factor-α |
| TRAF | Tumor necrosis factor receptor-associated factor |
| UBA | Ub-activating enzymes |
| UBC | Ub-conjugating enzymes |
| UCHs | Ubiquitin carboxy-terminal hydrolases |
| USPs | Ubiquitin-specific proteases |
References
- Han, S.; Mallampalli, R.K. The acute respiratory distress syndrome: From mechanism to translation. J. Immunol 2015, 194, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Matthay, M.A.; Song, Y.; Bai, C.; Jones, K.D. The acute respiratory distress syndrome in 2013. Transl. Respir. Med. 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Confalonieri, M.; Salton, F.; Fabiano, F. Acute respiratory distress syndrome. Eur. Respir. Rev. 2017, 26, 160116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Herold, S.; Gabrielli, N.M.; Vadasz, I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L665–L681. [Google Scholar] [CrossRef]
- Dada, L.A.; Sznajder, J.I. Mechanisms of pulmonary edema clearance during acute hypoxemic respiratory failure: Role of the Na,K-ATPase. Crit. Care Med. 2003, 31 (Suppl. 4), S248–S252. [Google Scholar] [CrossRef]
- Garcia, J.G.; Sznajder, J.I. Healthcare disparities in patients with acute respiratory distress syndrome. Toward Equity. Am. J. Respir Crit. Care Med. 2013, 188, 631–632. [Google Scholar] [CrossRef]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol 2011, 6, 147–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuckton, T.J.; Alonso, J.A.; Kallet, R.H.; Daniel, B.M.; Pittet, J.F.; Eisner, M.D.; Matthay, M.A. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N. Engl. J. Med. 2002, 346, 1281–1286. [Google Scholar] [CrossRef]
- Magnani, N.D.; Dada, L.A.; Sznajder, J.I. Ubiquitin-proteasome signaling in lung injury. Transl. Res. 2018, 198, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, I.; Weiss, C.H.; Sznajder, J.I. Ubiquitination and proteolysis in acute lung injury. Chest 2012, 141, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Helenius, I.T.; Dada, L.A.; Sznajder, J.I. Role of ubiquitination in Na,K-ATPase regulation during lung injury. Proc. Am. Thorac. Soc. 2010, 7, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Xu, C.; Kim, N.G.; Gumbiner, B.M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 2009, 8, 4032–4039. [Google Scholar] [CrossRef] [Green Version]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Weathington, N.M.; Sznajder, J.I.; Mallampalli, R.K. The emerging role of the ubiquitin proteasome in pulmonary biology and disease. Am. J. Respir Crit. Care Med. 2013, 188, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D. Ubiquitination and deubiquitination: Targeting of proteins for degradation by the proteasome. Semin. Cell Dev. Biol. 2000, 11, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbe, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [Green Version]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. NY Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Mialki, R.K.; Zhao, J.; Wei, J.; Mallampalli, D.F.; Zhao, Y. Overexpression of USP14 protease reduces I-kappaB protein levels and increases cytokine release in lung epithelial cells. J. Biol. Chem. 2013, 288, 15437–15441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Dong, S.; Bowser, R.K.; Khoo, A.; Zhang, L.; Jacko, A.M.; Zhao, Y.; Zhao, J. Regulation of the ubiquitylation and deubiquitylation of CREB-binding protein modulates histone acetylation and lung inflammation. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Guan, J.; Li, S.; Zhang, X.; Zheng, X. HSCARG downregulates NF-kappaB signaling by interacting with USP7 and inhibiting NEMO ubiquitination. Cell Death Dis. 2014, 5, e1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar]
- Zhao, J.; Wei, J.; Dong, S.; Bowser, R.K.; Zhang, L.; Jacko, A.M.; Zhao, Y. Destabilization of Lysophosphatidic Acid Receptor 1 Reduces Cytokine Release and Protects Against Lung Injury. EBioMedicine 2016, 10, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Bomberger, J.M.; Barnaby, R.L.; Stanton, B.A. The deubiquitinating enzyme USP10 regulates the endocytic recycling of CFTR in airway epithelial cells. Channels (Austin) 2010, 4, 150–154. [Google Scholar] [CrossRef] [Green Version]
- Palazon-Riquelme, P.; Worboys, J.D.; Green, J.; Valera, A.; Martin-Sanchez, F.; Pellegrini, C.; Brough, D.; Lopez-Castejon, G. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Colleran, A.; Collins, P.E.; O’Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of NF-kappaB by Ubiquitin-Specific Protease-7 promotes transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wei, J.; Li, S.; Jacko, A.M.; Weathington, N.M.; Mallampalli, R.K.; Zhao, J.; Zhao, Y. The deubiquitinase USP13 stabilizes the anti-inflammatory receptor IL-1R8/Sigirr to suppress lung inflammation. EBioMedicine 2019, 45, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liang, J.; Jiang, D.; Wang, C.; Dai, H. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir Res. 2015, 16, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, M.; Jing, Y.; Yin, X.; Ma, P.; Zhang, Z.; Wang, X.; Di, W.; Zhuang, G. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat. Commun 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Lim, J.H.; Jono, H.; Ha, U.H.; Xu, H.; Ishinaga, H.; Morino, S.; Xu, X.; Yan, C.; Kai, H.; et al. Tumor suppressor cylindromatosis acts as a negative regulator for Streptococcus pneumoniae-induced NFAT signaling. J. Biol. Chem. 2008, 283, 12546–12554. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhao, J.; Yu, J.; Zhang, W.; Huang, Y. Cylindromatosis (CYLD) inhibits Streptococcus pneumonia-induced plasminogen activator inhibitor-1 expression via interacting with TRAF-6. Biochem. Biophys. Res. Commun. 2015, 463, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Jono, H.; Kai, H.; Li, J.D. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J. Biol. Chem. 2005, 280, 41111–41121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Jono, H.; Koga, T.; Woo, C.H.; Ishinaga, H.; Bourne, P.; Xu, H.; Ha, U.H.; Xu, H.; Li, J.D. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS ONE 2007, 2, e1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Jono, H.; Komatsu, K.; Woo, C.H.; Lee, J.; Miyata, M.; Matsuno, T.; Xu, X.; Huang, Y.; Zhang, W.; et al. CYLD negatively regulates transforming growth factor-beta-signalling via deubiquitinating Akt. Nat. Commun. 2012, 3, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Stirling, B.; Derry, J.; Koga, T.; Jono, H.; Woo, C.H.; Xu, H.; Bourne, P.; Ha, U.H.; Ishinaga, H.; et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity 2007, 27, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Vidal, S.; El Motiam, A.; Seoane, R.; Preitakaite, V.; Bouzaher, Y.H.; Gomez-Medina, S.; San Martin, C.; Rodriguez, D.; Rejas, M.T.; Baz-Martinez, M.; et al. Regulation of the Ebola Virus VP24 Protein by SUMO. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Ali, A.; Raja, R.; Farooqui, S.R.; Ahmad, S.; Banerjea, A.C. USP7 deubiquitinase controls HIV-1 production by stabilizing Tat protein. Biochem. J. 2017, 474, 1653–1668. [Google Scholar] [CrossRef]
- Xiang, Q.; Ju, H.; Nicholas, J. USP7-Dependent Regulation of TRAF Activation and Signaling by a Viral Interferon Regulatory Factor Homologue. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomberger, J.M.; Ye, S.; Maceachran, D.P.; Koeppen, K.; Barnaby, R.L.; O’Toole, G.A.; Stanton, B.A. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 2011, 7, e1001325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, R.; Sugino, T.; Nolte, H.; Andrade, J.; Zimmermann, B.; Shi, C.; Doddaballapur, A.; Ong, Y.T.; Wilhelm, K.; Fasse, J.W.D.; et al. Deubiquitinase USP10 regulates Notch signaling in the endothelium. Science 2019, 364, 188–193. [Google Scholar] [PubMed]
- Wang, D.; Zhao, J.; Li, S.; Wei, J.; Nan, L.; Mallampalli, R.K.; Weathington, N.M.; Ma, H.; Zhao, Y. Phosphorylated E2F1 is stabilized by nuclear USP11 to drive Peg10 gene expression and activate lung epithelial cells. J. Mol. Cell Biol. 2018, 10, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.M.; Yu, C.Y.; Yang, H.C.; Ko, S.H.; Liao, C.L.; Lin, Y.L. Ubiquitin-specific protease 13 regulates IFN signaling by stabilizing STAT1. J. Immunol. 2013, 191, 3328–3336. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Q.; Jing, Y.Y.; Zhang, M.; Wang, H.Y.; Cai, Z.; Liuyu, T.; Zhang, Z.D.; Xiong, T.C.; Wu, Y.; et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Commun. 2017, 8, 15534. [Google Scholar] [CrossRef]
- Lu, Y.; Qiu, Y.; Chen, P.; Chang, H.; Guo, L.; Zhang, F.; Ma, L.; Zhang, C.; Zheng, X.; Xiao, J.; et al. ER-localized Hrd1 ubiquitinates and inactivates Usp15 to promote TLR4-induced inflammation during bacterial infection. Nat. Microbiol. 2019, 4, 2331–2346. [Google Scholar] [CrossRef]
- Song, H.; Tao, L.; Chen, C.; Pan, L.; Hao, J.; Ni, Y.; Li, D.; Li, B.; Shi, G. USP17-mediated deubiquitination and stabilization of HDAC2 in cigarette smoke extract-induced inflammation. Int. J. Clin. Exp. Pathol. 2015, 8, 10707–10715. [Google Scholar]
- Lu, C.H.; Yeh, D.W.; Lai, C.Y.; Liu, Y.L.; Huang, L.R.; Lee, A.Y.; Jin, S.C.; Chuang, T.H. USP17 mediates macrophage-promoted inflammation and stemness in lung cancer cells by regulating TRAF2/TRAF3 complex formation. Oncogene 2018, 37, 6327–6340. [Google Scholar] [CrossRef]
- Lei, C.Q.; Wu, X.; Zhong, X.; Jiang, L.; Zhong, B.; Shu, H.B. USP19 Inhibits TNF-alpha- and IL-1beta-Triggered NF-kappaB Activation by Deubiquitinating TAK1. J. Immunol. 2019, 203, 259–268. [Google Scholar] [CrossRef]
- Wu, X.; Lei, C.; Xia, T.; Zhong, X.; Yang, Q.; Shu, H.B. Regulation of TRIF-mediated innate immune response by K27-linked polyubiquitination and deubiquitination. Nat. Commun. 2019, 10, 4115. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jin, S.; Wang, R.F. The BECN1-USP19 axis plays a role in the crosstalk between autophagy and antiviral immune responses. Autophagy 2016, 12, 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Liu, X.; Wang, X.; Liu, X.; Li, H.; Darnay, B.G.; Lin, X.; Sun, S.C.; Dong, C. Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci Signal. 2013, 6, ra35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Wang, D.; Fang, L.; Zhang, H.; Luo, R.; Shang, M.; Ouyang, C.; Ouyang, H.; Chen, H.; Xiao, S. Ubiquitin-specific proteases 25 negatively regulates virus-induced type I interferon signaling. PLoS ONE 2013, 8, e80976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.; Zhang, M.; Zhang, M.X.; Ren, Y.; Jin, J.; Zhao, Q.; Pan, Z.; Wu, M.; Shu, H.B.; Dong, C.; et al. Induction of USP25 by viral infection promotes innate antiviral responses by mediating the stabilization of TRAF3 and TRAF6. Proc. Natl. Acad. Sci. USA 2015, 112, 11324–11329. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Liu, X.; Wang, X.; Chang, S.H.; Liu, X.; Wang, A.; Reynolds, J.M.; Dong, C. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat. Immunol. 2012, 13, 1110–1117. [Google Scholar] [CrossRef]
- Long, C.; Lai, Y.; Li, J.; Huang, J.; Zou, C. LPS promotes HBO1 stability via USP25 to modulate inflammatory gene transcription in THP-1 cells. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 773–782. [Google Scholar] [CrossRef]
- Long, C.; Lai, Y.; Li, T.; Nyunoya, T.; Zou, C. Cigarette smoke extract modulates Pseudomonas aeruginosa bacterial load via USP25/HDAC11 axis in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L252–L263. [Google Scholar] [CrossRef]
- Li, S.; Wang, D.; Zhao, J.; Weathington, N.M.; Shang, D.; Zhao, Y. The deubiquitinating enzyme USP48 stabilizes TRAF2 and reduces E-cadherin-mediated adherens junctions. FASEB J. 2018, 32, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Nan, L.; Jacko, A.M.; Tan, J.; Wang, D.; Zhao, J.; Kass, D.J.; Ma, H.; Zhao, Y. Ubiquitin carboxyl-terminal hydrolase-L5 promotes TGFbeta-1 signaling by de-ubiquitinating and stabilizing Smad2/Smad3 in pulmonary fibrosis. Sci. Rep. 2016, 6, 33116. [Google Scholar] [CrossRef] [Green Version]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215.e20–1230.e20. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Feng, Q.; Jin, L.; Huang, F.; Miao, Y.; Liu, J.; Xu, Y.; Chen, X.; Zhang, H.; Guo, T.; et al. Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat. Commun. 2020, 11, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zheng, H.; Mao, A.P.; Zhong, B.; Li, Y.; Liu, Y.; Gao, Y.; Ran, Y.; Tien, P.; Shu, H.B. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 2010, 285, 4291–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulas, F.; Wang, X.; Song, S.; Nishanth, G.; Yi, W.; Brunn, A.; Larsen, P.K.; Isermann, B.; Kalinke, U.; Barragan, A.; et al. The deubiquitinase OTUB1 augments NF-kappaB-dependent immune responses in dendritic cells in infection and inflammation by stabilizing UBC13. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yu, J.; Cheng, X.; Zhao, B.; Manyam, G.C.; Zhang, L.; Schluns, K.; Li, P.; Wang, J.; Sun, S.C. The deubiquitinase Otub1 controls the activation of CD8(+) T cells and NK cells by regulating IL-15-mediated priming. Nat. Immunol. 2019, 20, 879–889. [Google Scholar] [CrossRef]
- Edelmann, M.J.; Kramer, H.B.; Altun, M.; Kessler, B.M. Post-translational modification of the deubiquitinating enzyme otubain 1 modulates active RhoA levels and susceptibility to Yersinia invasion. FEBS J. 2010, 277, 2515–2530. [Google Scholar] [CrossRef]
- Jahan, A.S.; Biquand, E.; Munoz-Moreno, R.; Le Quang, A.; Mok, C.K.; Wong, H.H.; Teo, Q.W.; Valkenburg, S.A.; Chin, A.W.H.; Man Poon, L.L.; et al. OTUB1 Is a Key Regulator of RIG-I-Dependent Immune Signaling and Is Targeted for Proteasomal Degradation by Influenza A NS1. Cell Rep. 2020, 30, 1570.e6–1584.e6. [Google Scholar] [CrossRef]
- Herhaus, L.; Al-Salihi, M.; Macartney, T.; Weidlich, S.; Sapkota, G.P. OTUB1 enhances TGFbeta signalling by inhibiting the ubiquitylation and degradation of active SMAD2/3. Nat. Commun. 2013, 4, 2519. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, J.; Qian, L.; Feng, Q.; Wang, X.; Yuan, Y.; Zuo, Y.; Cheng, Q.; Miao, Y.; Guo, T.; et al. Induction of OTUD1 by RNA viruses potently inhibits innate immune responses by promoting degradation of the MAVS/TRAF3/TRAF6 signalosome. PLoS Pathog. 2018, 14, e1007067. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Song, J.; Sun, Y.; Qi, F.; Liu, L.; Jin, Y.; McNutt, M.A.; Yin, Y. Mutations of deubiquitinase OTUD1 are associated with autoimmune disorders. J. Autoimmun. 2018, 94, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, D.; Wang, P.; Zhao, Y.; You, F. OTUD1 Negatively Regulates Type I IFN Induction by Disrupting Noncanonical Ubiquitination of IRF3. J. Immunol. 2020, 204, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mudge, M.C.; Soll, J.M.; Rodrigues, R.B.; Byrum, A.K.; Schwarzkopf, E.A.; Bradstreet, T.R.; Gygi, S.P.; Edelson, B.T.; Mosammaparast, N. OTUD4 Is a Phospho-Activated K63 Deubiquitinase that Regulates MyD88-Dependent Signaling. Mol. Cell. 2018, 69, 505.e5–516.e5. [Google Scholar] [CrossRef] [Green Version]
- Liuyu, T.; Yu, K.; Ye, L.; Zhang, Z.; Zhang, M.; Ren, Y.; Cai, Z.; Zhu, Q.; Lin, D.; Zhong, B. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res. 2019, 29, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Majid, M.C.; Soll, J.M.; Brickner, J.R.; Dango, S.; Mosammaparast, N. Noncanonical regulation of alkylation damage resistance by the OTUD4 deubiquitinase. EMBO J. 2015, 34, 1687–1703. [Google Scholar] [CrossRef] [Green Version]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Ahmad, R.C.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O.; et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004, 5, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Ma, A.; Harhaj, E.W. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010, 327, 1135–1139. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Jia, Y.; Han, S.; Wang, X.; Han, F.; Zhang, J.; Zhang, W.; Guan, H.; Hu, D. Klf4 Alleviates Lipopolysaccharide-Induced Inflammation by Inducing Expression of MCP-1 Induced Protein 1 to Deubiquitinate TRAF6. Cell Physiol. Biochem. 2018, 47, 2278–2290. [Google Scholar] [CrossRef]
- Feng, Q.; Miao, Y.; Ge, J.; Yuan, Y.; Zuo, Y.; Qian, L.; Liu, J.; Cheng, Q.; Guo, T.; Zhang, L.; et al. ATXN3 Positively Regulates Type I IFN Antiviral Response by Deubiquitinating and Stabilizing HDAC3. J. Immunol. 2018, 201, 675–687. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. JOSD1 inhibits mitochondrial apoptotic signalling to drive acquired chemoresistance in gynaecological cancer by stabilizing MCL1. Cell Death Differ. 2020, 27, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Zhang, Y.; Zhao, P.; Qian, L.; Yuan, Y.; Liu, J.; Cheng, Q.; Xu, W.; Zuo, Y.; et al. JOSD1 Negatively Regulates Type-I Interferon Antiviral Activity by Deubiquitinating and Stabilizing SOCS1. Viral Immunol. 2017, 30, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Wang, B.; Xu, G.; Yang, Z.; Tang, M.; Ma, A.; Jing, T.; Xu, X.; Zhang, X.; et al. POH1 deubiquitinates pro-interleukin-1beta and restricts inflammasome activity. Nat. Commun. 2018, 9, 4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednash, J.S.; Weathington, N.; Londino, J.; Rojas, M.; Gulick, D.L.; Fort, R.; Han, S.; McKelvey, A.C.; Chen, B.B.; Mallampalli, R.K. Targeting the deubiquitinase STAMBP inhibits NALP7 inflammasome activity. Nat. Commun. 2017, 8, 15203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Duan, M.; Hibbs, M.L.; Chen, W. The contributions of lung macrophage and monocyte heterogeneity to influenza pathogenesis. Immunol. Cell Biol. 2017, 95, 225–235. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185.e6–197.e6. [Google Scholar] [CrossRef] [Green Version]
- Brummelkamp, T.R.; Nijman, S.M.; Dirac, A.M.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef]
- Cai, J.; Culley, M.K.; Zhao, Y.; Zhao, J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell 2018, 9, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.Y. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (COVID-19) Pneumonia in Two Patients with Lung Cancer. J. Thorac. Oncol. 2020, 15, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, P1517–P1520. [Google Scholar] [CrossRef]
- Bekes, M.; Rut, W.; Kasperkiewicz, P.; Mulder, M.P.; Ovaa, H.; Drag, M.; Lima, C.D.; Huang, T.T. SARS hCoV papain-like protease is a unique Lys48 linkage-specific di-distributive deubiquitinating enzyme. Biochem. J. 2015, 468, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Garcia, G.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes are Susceptible to SARS-CoV-2 Infection. bioRxiv 2020. [Google Scholar]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016.e19–1035.e19. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020. [Google Scholar]
- Renatus, M.; Parrado, S.G.; D’Arcy, A.; Eidhoff, U.; Gerhartz, B.; Hassiepen, U.; Pierrat, B.; Riedl, R.; Vinzenz, D.; Worpenberg, S.; et al. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure 2006, 14, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Finerty, P.J., Jr.; Mackenzie, F.; Newman, E.M.; Dhe-Paganon, S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8). J. Biol. Chem. 2006, 281, 38061–38070. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chenova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R. CYLD: A deubiquitination enzyme with multiple roles in cancer. Future Oncol. 2011, 7, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israel, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef]
- Jono, H.; Lim, J.H.; Chen, L.F.; Xu, H.; Trompouki, E.; Pan, Z.K.; Mosialos, G.; Li, J.D. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: Evidence for a novel inducible autoregulatory feedback pathway. J. Biol. Chem. 2004, 279, 36171–36174. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Ha, U.H.; Woo, C.H.; Xu, H.; Li, J.D. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cell Microbiol. 2008, 10, 2247–2256. [Google Scholar] [CrossRef]
- Daviet, L.; Colland, F. Targeting ubiquitin specific proteases for drug discovery. Biochimie 2008, 90, 270–283. [Google Scholar] [CrossRef]
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.M.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun 2014, 5, 4763. [Google Scholar] [CrossRef] [Green Version]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Hacker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.K.; Pietras, E.M.; He, J.Q.; Kang, J.R.; Liu, S.Y.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.L.; Wang, Y.; et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006, 25, 3257–3263. [Google Scholar] [CrossRef]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A deubiquitinase that regulates type I interferon production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Li, F.; Long, Y.; Zheng, J. Chloroquine attenuates lipopolysaccharide-induced inflammatory responses through upregulation of USP25. Can. J. Physiol. Pharmacol. 2017, 95, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.; Hospenthal, M.K.; Geurink, P.P.; Elliott, P.R.; Akutsu, M.; Arnaudo, N.; Ekkebus, R.; Kulathu, Y.; Wauer, T.; El Oualid, F.; et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 2013, 154, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, F. Linear ubiquitination signals in adaptive immune responses. Immunol. Rev. 2015, 266, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Fiil, B.K.; Gyrd-Hansen, M. OTULIN deficiency causes auto-inflammatory syndrome. Cell Res. 2016, 26, 1176–1177. [Google Scholar] [CrossRef] [Green Version]
- Juang, Y.C.; Landry, M.C.; Sanches, M.; Vittal, V.; Leung, C.C.; Ceccarelli, D.F.; Mateo, A.R.; Pruneda, J.N.; Mao, D.Y.; Szilard, R.K.; et al. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol. Cell 2012, 45, 384–397. [Google Scholar] [CrossRef] [Green Version]
- Nakada, S.; Tai, I.; Panier, S.; Al-Hakim, A.; Iemura, S.; Juang, Y.C.; O’Donnell, L.; Kumakubo, A.; Munro, M.; Sicheri, F.; et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 2010, 466, 941–946. [Google Scholar] [CrossRef]
- Wang, T.; Yin, L.; Cooper, E.M.; Lai, M.Y.; Dickey, S.; Pickart, C.M.; Fushman, D.; Wilkinson, K.D.; Cohen, R.E.; Wolberger, C. Evidence for bidentate substrate binding as the basis for the K48 linkage specificity of otubain 1. J. Mol. Biol. 2009, 386, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Wiener, R.; Zhang, X.; Wang, T.; Wolberger, C. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 2012, 483, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matmati, M.; Jacques, P.; Maelfait, J.; Verheugen, E.; Kool, M.; Sze, M.; Geboes, L.; Louagie, E.; Mc Guire, C.; Vereecke, L.; et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 2011, 43, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Liu, J.; Wu, X.; Liu, S.; Li, G.; Han, C.; Song, L.; Li, Z.; Wang, Q.; Wang, J.; et al. Histone methyltransferase Ash1l suppresses interleukin-6 production and inflammatory autoimmune diseases by inducing the ubiquitin-editing enzyme A20. Immunity 2013, 39, 470–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinojosa, C.A.; Akula Suresh Babu, R.; Rahman, M.M.; Fernandes, G.; Boyd, A.R.; Orihuela, C.J. Elevated A20 contributes to age-dependent macrophage dysfunction in the lungs. Exp. Gerontol. 2014, 54, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Ma, A.; Zhang, L.; Jin, W.L.; Qian, Y.; Xu, G.; Qiu, B.; Yang, Z.; Liu, Y.; Xia, Q.; et al. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nat. Commun. 2015, 6, 8704. [Google Scholar] [CrossRef]
- Cooper, E.M.; Cutcliffe, C.; Kristiansen, T.Z.; Pandey, A.; Pickart, C.M.; Cohen, R.E. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009, 28, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Yoshikawa, A.; Yamagata, A.; Mimura, H.; Yamashita, M.; Ookata, K.; Nureki, O.; Iwai, K.; Komada, M.; Fukai, S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature 2008, 455, 358–362. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Mallery, D.L.; Watkinson, R.E.; Dickson, C.F.; James, L.C. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc. Natl. Acad. Sci. USA 2015, 112, 10014–10019. [Google Scholar] [CrossRef] [Green Version]
- McCullough, J.; Row, P.E.; Lorenzo, O.; Doherty, M.; Beynon, R.; Clague, M.J.; Urbe, S. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr. Biol. 2006, 16, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Wang, J.; Chen, J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J. Biol. Chem. 2010, 285, 30982–30988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carolan, B.J.; Heguy, A.; Harvey, B.G.; Leopold, P.L.; Ferris, B.; Crystal, R.G. Up-regulation of expression of the ubiquitin carboxyl-terminal hydrolase L1 gene in human airway epithelium of cigarette smokers. Cancer Res. 2006, 66, 10729–10740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Shen, X. Ubiquitin carboxyl-terminal hydrolases: Involvement in cancer progression and clinical implications. Cancer Metastasis Rev. 2017, 36, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Niu, X.; Li, Z.; Yu, Y.; Ye, X.; Lu, S.; Chen, Z. Effect of ubiquitin carboxy-terminal hydrolase 37 on apoptotic in A549 cells. Cell Biochem. Funct. 2011, 29, 142–148. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Niu, J.; Azfer, A.; Zhelyabovska, O.; Fatma, S.; Kolattukudy, P.E. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J. Biol. Chem. 2008, 283, 14542–14551. [Google Scholar] [CrossRef] [Green Version]
- Skalniak, L.; Mizgalska, D.; Zarebski, A.; Wyrzykowska, P.; Koj, A.; Jura, J. Regulatory feedback loop between NF-kappaB and MCP-1-induced protein 1 RNase. FEBS J. 2009, 276, 5892–5905. [Google Scholar] [CrossRef]
- Huang, S.; Miao, R.; Zhou, Z.; Wang, T.; Liu, J.; Liu, G.; Chen, Y.E.; Xin, H.B.; Zhang, J.; Fu, M. MCPIP1 negatively regulates toll-like receptor 4 signaling and protects mice from LPS-induced septic shock. Cell Signal. 2013, 25, 1228–1234. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Wang, J.; Azfer, A.; Song, W.; Tromp, G.; Kolattukudy, P.E.; Fu, M. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J. Biol. Chem. 2008, 283, 6337–6346. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Saad, Y.; Lei, T.; Wang, J.; Qi, D.; Yang, Q.; Kolattukudy, P.E.; Fu, M. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J. Exp. Med. 2010, 207, 2959–2973. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Chien, H.L.; Lin, S.Y.; Chang, B.L.; Yu, H.P.; Tang, W.C.; Lin, Y.L. MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res. 2013, 41, 3314–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruneda, J.N.; Durkin, C.H.; Geurink, P.P.; Ovaa, H.; Santhanam, B.; Holden, D.W.; Komander, D. The Molecular Basis for Ubiquitin and Ubiquitin-like Specificities in Bacterial Effector Proteases. Mol. Cell 2016, 63, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey-Elkin, B.A.; Knaap, R.C.; Johnson, G.G.; Dalebout, T.J.; Ninaber, D.K.; van Kasteren, P.B.; Bredenbeek, P.J.; Snijder, E.J.; Kikkert, M.; Mark, B.L. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014, 289, 34667–34682. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Mesters, J.R.; Drosten, C.; Anemuller, S.; Ma, Q.; Hilgenfeld, R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antiviral Res. 2014, 109, 72–82. [Google Scholar] [CrossRef]
- Frias-Staheli, N.; Giannakopoulos, N.V.; Kikkert, M.; Taylor, S.L.; Bridgen, A.; Paragas, J.; Richt, J.A.; Rowland, R.R.; Schmaljohn, C.S.; Lenschow, D.J.; et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2007, 2, 404–416. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| DUBs | Target Genes | Function |
|---|---|---|
| CYLD | TAK1 [43] | Negatively regulates S.p. induced NFAT signaling [43] |
| TRAF6 [44] | Inhibits S.p. induced PAI-1 expression [44] | |
| TRAF6/TRAF7 [45,46] | Regulates TLR4 signaling [45] Inhibits inflammation [46] | |
| AKT [47] | Regulates TGF- β signaling [47] | |
| PAI-1 [48] | Regulates acute lung injury [48] | |
| USP-7 | NLRP3 [38] | Regulates NLRP3 inflammasome activation [38] |
| NF-κB [39], NEMO [33] | Regulates NF-κB signaling [33,39] | |
| VP24 [49] | Involves in virus replication [49] | |
| Tat [50] | Involves in virus production [50] | |
| TRAF3/TRAF6 [51] | Modulates antiviral signaling [51] | |
| TRAF6/IKKγ [34] | Regulates TLR signaling [34] | |
| USP-10 | CFTR [37,52] | Epithelial mucosal clearance [37,52] |
| NICD1 [53] | Regulates Notch signaling [53] | |
| USP-11 | E2F1 [54] | Regulates lung epithelia proliferation and wound healing [54] |
| LPA1 [36] | Enhances inflammation [36] | |
| USP-13 | IL-1R8/Sigirr [40] | Suppresses lung inflammation [40] |
| PTEN [41] | Regulates cell apoptosis [41] | |
| MCL1 [42] | Regulates transformation of fibroblasts [42] | |
| STAT1 [55] | Regulates IFN Signaling [55] | |
| STING [56] | Negatively regulates antiviral responses [56] | |
| USP-14 | I-kB [31] | Increases cytokine release [31] |
| CBP [32] | Lung inflammation [32] | |
| USP-15 | IκBα [57] | NF-κB activation [57] |
| USP-17 | HDAC2 [58] | Reverses glucocorticoid resistance [58] |
| TRAF2/TRAF3 [59] | Lung inflammation [59] | |
| USP-19 | TAK1 [60] | Inhibits NF-κB activation [60] |
| TRIF [61] | Inactivates TLR3/4-mediated innate immune responses [61] | |
| BECN1 [62] | Promotes formation of autophagosomes and inhibits DDX58/RIG-I-mediated type I interferon signaling [62] | |
| USP-25 | TRAF3 [63] | Regulates TLR4-dependent Innate Immune Responses [63] |
| RIG-I/TRAF2/TRAF6 [64] | Negatively regulates virus-induced type I interferon signaling [64] | |
| TRAF3/TRAF6 [65] | Promotes Innate Antiviral Responses [65] | |
| TRAF5 and TRAF6 [66] | Regulates IL-17 signaling [66] | |
| HBO1 [67] | Modulates macrophage inflammation [67] | |
| HDAC11 [68] | Modulates bacteria load [68] | |
| USP-48 | TRAF2 [69] | Reduces E-cadherin-mediated adherence junctions [69] |
| UCHL5(UCH37) | Smad2/Smad3 [70] | Promotes TGFβ-1 signaling [70] |
| OTULIN | Met-1 [71,72,73] | Prevents inflammation [71,72,73] |
| STAT1 [74] | Controls antiviral signaling [74] | |
| OTUB1 | TRAF3/TRAF6 [75] | Negatively regulates virus-triggered type I IFN induction [75] |
| UBC13 [76] | Augments NF-κB-dependent Immune Responses [76] | |
| AKT [77] | Controls the activation of CD8 + T Cells and NK Cells [77] | |
| RhoA [78] | Increases bacteria uptake [78] | |
| RIG-1 [79] | Activates RIG-I signaling cascade and antiviral responses [79] | |
| Smad2/3 [80] | Enhances TGFβ signaling [80] | |
| OTUD1 | MAVS/TRAF3/TRAF6 [81] | Inhibits Innate Immune Responses [81] |
| IRF3 [82,83] | Maintains immune homeostasis [82] Negatively regulates Type I IFN induction [83] | |
| OTUD4 | MyD88 [84] | Suppresses TLR signaling [84] |
| MAVS [85] | Regulates innate antiviral responses [85] | |
| ALKBH3 [86] | Regulates DNA damage [86] | |
| A20 | TRAF6 [87] | Restricts TLR signals [87] |
| TRAF2/TRAF6/Ubc13/UbcH5c [88] | Inhibits NF-kappa B Signaling [88] | |
| MCPIP1 | TRAF6 [89] | Impedes NF-κB and inflammatory signaling [89] |
| ATXN3 | HDAC3 [90] | Positively regulates type I IFN antiviral response [90] |
| JOSD1 | MCL [91] | Inhibits mitochondrial apoptotic signaling [91] |
| SOCS1 [92] | Inhibits type I IFN signaling and antiviral response [92] | |
| POH1 | pro-IL-1β [93] | Negatively regulates the immune response [93] |
| BRCC3 | NLRP3 [94] | Promotes the inflammasome activation [94] |
| STAMBP | NALP7 [95] | Reduces pro-inflammatory stress [95] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zou, C. The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 4842. https://doi.org/10.3390/ijms21144842
Li T, Zou C. The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2020; 21(14):4842. https://doi.org/10.3390/ijms21144842
Chicago/Turabian StyleLi, Tiao, and Chunbin Zou. 2020. "The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 21, no. 14: 4842. https://doi.org/10.3390/ijms21144842
APA StyleLi, T., & Zou, C. (2020). The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences, 21(14), 4842. https://doi.org/10.3390/ijms21144842