Carotid Body and Metabolic Syndrome: Mechanisms and Potential Therapeutic Targets

Abstract
:1. Introduction
2. Hypoxia and Metabolic Dysfunction: What Is the Role of CB?
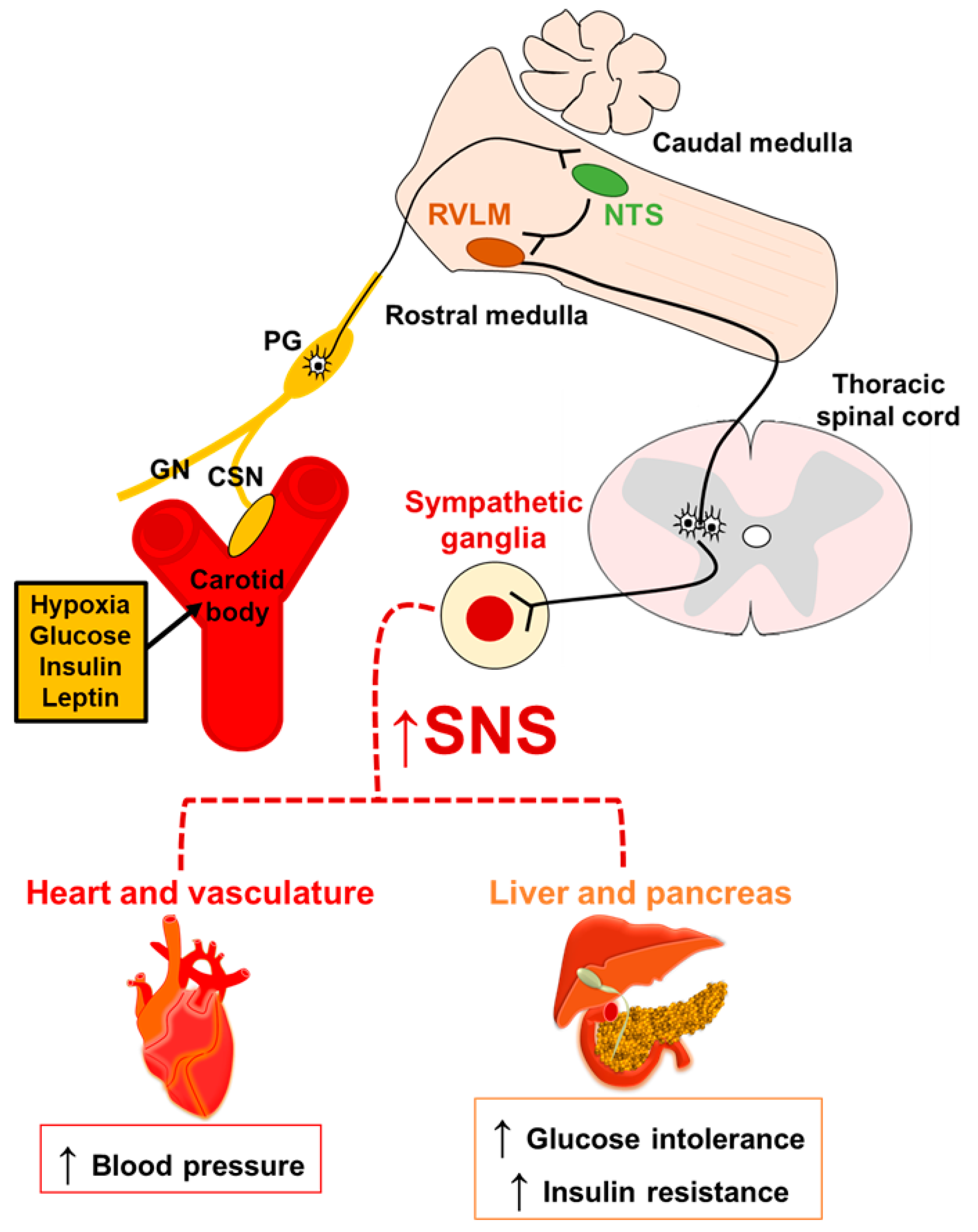
3. CB and Sympathetic Activity: A Common Way to Metabolic Dysfunction
4. Metabolic Syndrome and CB Chemosensory Response
4.1. Hypertension and CB Chemoreflex
4.2. Glucose Metabolism and CB Chemoreflex
4.2.1. Does CB Directly Sense Glucose Levels?
4.2.2. Insulin: A Better Marker of CB-Induced Metabolic Dysfunctions?
4.3. CB Chemoreflex in Obesity: How to Dissociate the Metabolic Effects
Obesity, Leptin and OSA: A Trio for CB Activation
5. CB Manipulations: Therapeutic Targets for Metabolic Syndrome
5.1. CB Resection: Do the Benefits Outweigh the Costs?
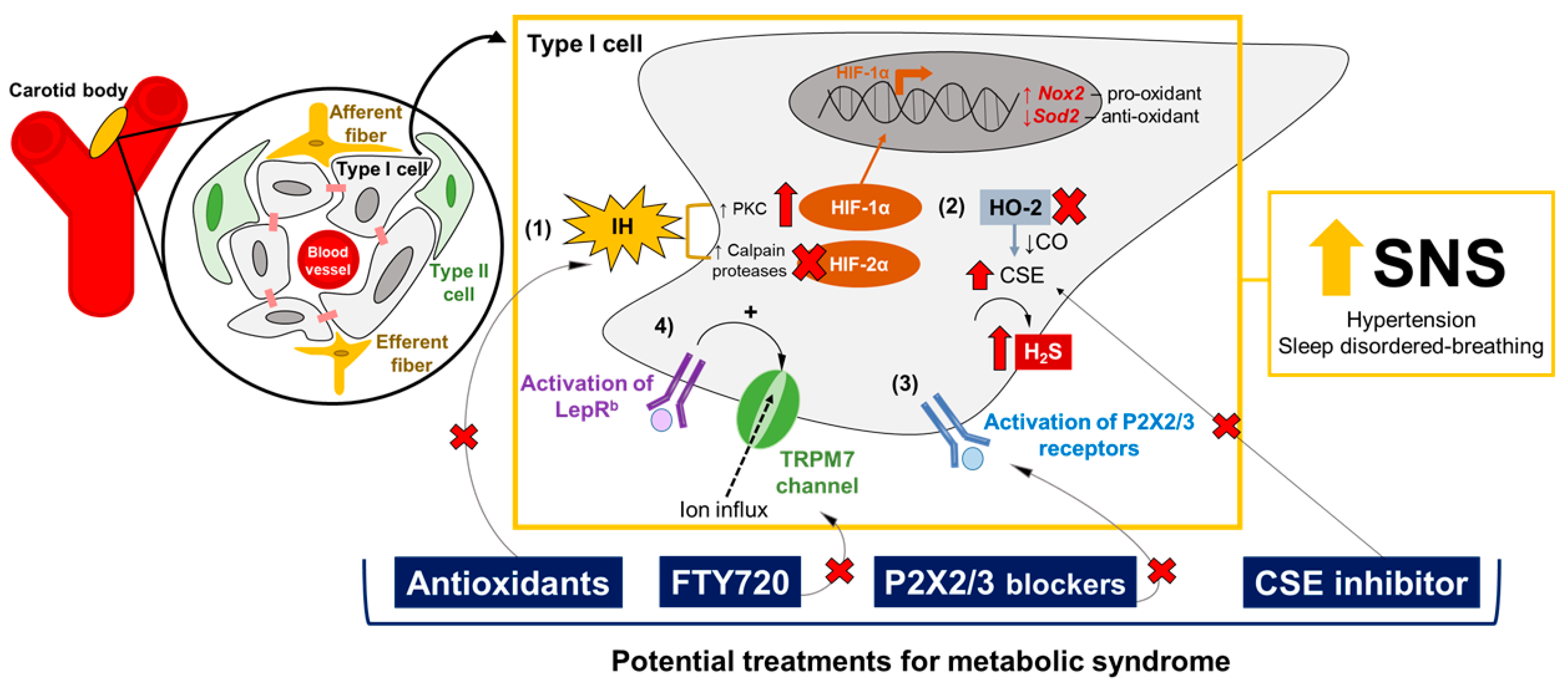
5.2. Molecular Targets in the CB: Promising Pharmacology for Metabolic Syndrome
- -
- HIF-1α and HIF-2α pathways: Dr. Gregg Semenza’s and Dr. Nanduri Prabhakar’s laboratories have provided plenty of evidence that ROS are involved in the overactivation of CB induced by IH [55,99,193,194]. IH augments the production of ROS in the CB, especially increasing the superoxide anion levels and the consequent elevation of hydrogen peroxide (H2O2) [195,196]. IH-induced ROS production in the CB occurs through different mechanisms, such as the activation of NADPH oxidase 2 [196] and the inhibition of superoxide dismutase 2 (Sod2) [197]. The transcriptional regulation of IH-induced oxidative stress in the CB is mainly governed by the balance between HIF-1α and HIF-2α signaling [99,193,194,198]. Both are heterodimeric transcription factors involved in oxygen homeostasis [198], and are expressed in the CB’s glomus cells [199]. However, HIF-1α and HIF-2α have opposite functions in the CB: HIF-1α activates Nox2 gene expression, the gene encoding to the pro-oxidant enzyme NADPH oxidase 2, while HIF-2α promotes the transcription of the Sod2 gene, inducing the expression of the Sod2 enzyme responsible for catalyzing the conversion of superoxide to hydrogen peroxide [193,194,197,198]. IH increases HIF-1α levels, and induces the degradation of HIF-2α via Ca+2-dependent protein kinase C (PKC) and calpains proteases, respectively [197,200]. Pharmacological and genetic manipulations of the HIF-1α and HIF-2α pathways in the CB have shown promising effects on the regulation of the CB’s chemoreflex, ventilatory stability and blood pressure [201,202] (Table 1), through a mechanism of mutual antagonism [203]. Therefore, drugs that selectively inhibit HIF-1α or upregulate HIF-2α in the CB, modulating the expression of NADPH oxidase 2 and Sod2 enzymes, could be potential targets for hypertension and OSA.
- -
- Gasotransmitters: Dr. Nanduri Prabhakar’s laboratory have shown that CO and H2S may mediate the CB’s chemosensory response to hypoxia [194,204]. CO, which is generated by hemeoxygenase-2 (HO-2), inhibits CB activity [205], while H2S is catalyzed by the enzyme CSE and stimulates the hypoxic response in the CB [204]. The pharmacological blockade of CSE reduces H2S levels and normalizes breathing and blood pressure [53,204,206] (Table 1). Therefore, targeting the upregulation of HO-2 and the downregulation of CSE, with consequent increases of CO and the reduction of H2S generation in the CB, may be a pharmacological intervention for metabolic syndrome, attenuating hypertension and OSA. Moreover, Yuan and collaborators [207] have shown that IH-evoked ROS inactivates HO-2 in the CB, increasing the generation of H2S, suggesting that the balance of gasotransmitters could also be involved in the HIF-1α and HIF-2α signaling pathways in the CB [194].
- -
- Purinergic receptors: The peripheral chemoreflex involves multiple excitatory postsynaptic responses, and ATP is the main neurotransmitter responsible for the activation of petrosal chemoreceptive terminals by binding to P2X2/3 receptors [208,209,210]. P2X2/3 receptors are also expressed in the glomus cells, promoting the excitation of the CB units induced by hypoxia and hypercapnia [211]. Dr. Julian Paton’s laboratory has shown that purinergic signaling may play a crucial role in the generation of aberrant chemoreflex responses in the CB, leading to hypertension and sleep-disordered breathing. Hence, the pharmacological antagonism of purinergic receptors in the CB has been proposed as a potential pharmacological approach to normalizing blood pressure and breathing stability [212,213] (Table 1).
- -
- Leptin-mediated TRPM7 channels: Our group has shown that leptin acts in the CB and increases CSN activity to increase blood pressure through the activation of TRPM7 channels [96,97]. Considering that leptin exerts multiple functions, regulating the metabolic rate and energy expenditure [154,155,156], and that leptin resistance is often observed in obese patients [13,16,17], we propose that the pharmacological blockade of the TRPM7 channels in the CB could be a potential and more feasible therapy for metabolic syndrome. In this scenario, the administration of FTY720 to the CB, a potent inhibitor of the TRPM7 channels, has shown promising effects on the control of blood pressure [96] (Table 1). Our group has also demonstrated that leptin is a potent stimulator of ventilation and HVR, via its activating of the CB’s glomus cells as well as CSN activity [98]. Thus, we hypothesized that the blockade of TRPM7 channels with FTY720 in the CB could also be a treatment for sleep-disordered breathing (Table 1).
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ford, E.S.; Giles, W.H.; Dietz, W.H. Prevalence of the metabolic syndrome among US adults: Findings from the Third National Health and Nutrition Examination Survey. J. Am. Med. Assoc. 2002, 287, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Sánchez, H.; Harhay, M.O.; Harhay, M.M.; McElligott, S. Prevalence and Trends of Metabolic Syndrome in the Adult U.S. Population, 1999–2010. J. Am. Coll. Cardiol. 2013, 62, 697–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.X.; Chaudhary, N.; Akinyemiju, T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev. Chronic Dis. 2017, 14, E24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Galassi, A.; Reynolds, K.; He, J. Metabolic syndrome and risk of cardiovascular disease: A meta-analysis. Am. J. Med. 2006, 119, 812–819. [Google Scholar] [CrossRef]
- Tentolouris, N.; Liatis, S.; Katsilambros, N. Sympathetic system activity in obesity and metabolic syndrome. Ann. N. Y. Acad. Sci. 2006, 1083, 129–152. [Google Scholar] [CrossRef]
- Conde, S.V.; Sacramento, J.F.; Guarino, M.P. Carotid body: A metabolic sensor implicated in insulin resistance. Physiol. Genom. 2018, 50, 208–214. [Google Scholar] [CrossRef]
- Baskin, D.G.; Stein, L.J.; Ikeda, H.; Woods, S.C.; Figlewicz, D.P.; Porte, D.; Greenwood, M.R.; Dorsa, D.M. Genetically obese Zucker rats have abnormally low brain insulin content. Life Sci. 1985, 36, 627–633. [Google Scholar] [CrossRef]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Peskind, E.; Raskind, M.; Boyko, E.J.; Porte, D. Cerebrospinal fluid leptin levels: Relationship to plasma levels and to adiposity in humans. Nat. Med. 1996, 2, 589–593. [Google Scholar] [CrossRef]
- Kaiyala, K.J.; Prigeon, R.L.; Kahn, S.E.; Woods, S.C.; Schwartz, M.W. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 2000, 49, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Urayama, A.; Banks, W.A. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology 2008, 149, 3592–3597. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Rui, L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1247–E1259. [Google Scholar] [CrossRef] [Green Version]
- Scarpace, P.J.; Zhang, Y. Leptin resistance: A prediposing factor for diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R493–R500. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Sáenz, P.; López-Barneo, J. Physiology of the Carotid Body: From Molecules to Disease. Annu. Rev. Physiol. 2020, 82, 127–149. [Google Scholar] [CrossRef] [Green Version]
- Nurse, C.A. Synaptic and paracrine mechanisms at carotid body arterial chemoreceptors. J. Physiol. 2014, 592, 3419–3426. [Google Scholar] [CrossRef]
- Platero-Luengo, A.; González-Granero, S.; Durán, R.; Díaz-Castro, B.; Piruat, J.I.; García-Verdugo, J.M.; Pardal, R.; López-Barneo, J. An O2-sensitive glomus cell-stem cell synapse induces carotid body growth in chronic hypoxia. Cell 2014, 156, 291–303. [Google Scholar] [CrossRef] [Green Version]
- López-Barneo, J.; Ortega-Sáenz, P.; González-Rodríguez, P.; Fernández-Agüera, M.C.; Macías, D.; Pardal, R.; Gao, L. Oxygen-sensing by arterial chemoreceptors: Mechanisms and medical translation. Mol. Asp. Med. 2016, 47, 90–108. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martín, M.C.; Vega-Agapito, M.V.; Conde, S.V.; Castañeda, J.; Bustamante, R.; Olea, E.; Perez-Vizcaino, F.; Gonzalez, C.; Obeso, A. Carotid body function and ventilatory responses in intermittent hypoxia. evidence for anomalous brainstem integration of arterial chemoreceptor input. J. Cell. Physiol. 2011, 226, 1961–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.Q.; Schreihofer, A.M. Altered sympathetic reflexes and vascular reactivity in rats after exposure to chronic intermittent hypoxia. J. Physiol. 2011, 589, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Kumar, G.K.; Peng, Y.-J. Sympatho-adrenal activation by chronic intermittent hypoxia. J. Appl. Physiol. 2012, 113, 1304–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurse, C.A.; Piskuric, N.A. Signal processing at mammalian carotid body chemoreceptors. Semin. Cell Dev. Biol. 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R. Sensing hypoxia: Physiology, genetics and epigenetics. J. Physiol. 2013, 591, 2245–2257. [Google Scholar] [CrossRef]
- Conde, S.V.; Sacramento, J.F.; Guarino, M.P.; Gonzalez, C.; Obeso, A.; Diogo, L.N.; Monteiro, E.C.; Ribeiro, M.J. Carotid body, insulin, and metabolic diseases: Unraveling the links. Front. Physiol. 2014, 5, 418. [Google Scholar] [CrossRef] [Green Version]
- Iturriaga, R.; Del Rio, R.; Idiaquez, J.; Somers, V.K. Carotid body chemoreceptors, sympathetic neural activation, and cardiometabolic disease. Biol. Res. 2016, 49, 13. [Google Scholar] [CrossRef] [Green Version]
- Conde, S.V.; Ribeiro, M.J.; Melo, B.F.; Guarino, M.P.; Sacramento, J.F. Insulin resistance: A new consequence of altered carotid body chemoreflex? J. Physiol. 2017, 595, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Chua, T.P.; Anker, S.D.; Francis, D.P.; Doehner, W.; Banasiak, W.; Poole-Wilson, P.A.; Piepoli, M.F.; Coats, A.J. Peripheral chemoreceptor hypersensitivity: An ominous sign in patients with chronic heart failure. Circulation 2001, 104, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Chien, M.-S.; Kaleem, S.; Matsunami, H. Single cell transcriptome analysis of mouse carotid body glomus cells. J. Physiol. 2016, 594, 4225–4251. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Prabhakar, N.R. Peripheral chemoreceptors: Function and plasticity of the carotid body. Compr. Physiol. 2012, 2, 141–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, V.; Pequignot, J.-M. Breathing at high altitude. Cell. Mol. Life Sci. 2009, 66, 3565–3573. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Dick, T.E.; Nanduri, J.; Kumar, G.K. Systemic, cellular and molecular analysis of chemoreflex-mediated sympathoexcitation by chronic intermittent hypoxia. Exp. Physiol. 2007, 92, 39–44. [Google Scholar] [CrossRef]
- Dempsey, J.A.; Veasey, S.C.; Morgan, B.J.; O’Donnell, C.P. Pathophysiology of sleep apnea. Physiol. Rev. 2010, 90, 47–112. [Google Scholar] [CrossRef]
- Jun, J.; Polotsky, V.Y. Metabolic consequences of sleep-disordered breathing. ILAR J. 2009, 50, 289–306. [Google Scholar] [CrossRef] [Green Version]
- Drager, L.F.; Togeiro, S.M.; Polotsky, V.Y.; Lorenzi-Filho, G. Obstructive sleep apnea: A cardiometabolic risk in obesity and the metabolic syndrome. J. Am. Coll. Cardiol. 2013, 62, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Iiyori, N.; Alonso, L.C.; Li, J.; Sanders, M.H.; Garcia-Ocana, A.; O’Doherty, R.M.; Polotsky, V.Y.; O’Donnell, C.P. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am. J. Respir. Crit. Care Med. 2007, 175, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Polotsky, V.Y.; Li, J.; Punjabi, N.M.; Rubin, A.E.; Smith, P.L.; Schwartz, A.R.; O’Donnell, C.P. Intermittent hypoxia increases insulin resistance in genetically obese mice. J. Physiol. 2003, 552, 253–264. [Google Scholar] [CrossRef]
- Drager, L.F.; Li, J.; Reinke, C.; Bevans-Fonti, S.; Jun, J.C.; Polotsky, V.Y. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obesity 2011, 19, 2167–2174. [Google Scholar] [CrossRef]
- Jun, J.C.; Shin, M.-K.; Devera, R.; Yao, Q.; Mesarwi, O.; Bevans-Fonti, S.; Polotsky, V.Y. Intermittent hypoxia-induced glucose intolerance is abolished by α-adrenergic blockade or adrenal medullectomy. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1073–E1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.-K.; Han, W.; Joo, H.; Bevans-Fonti, S.; Shiota, M.; Stefanovski, D.; Polotsky, V.Y. Effect of adrenal medullectomy on metabolic responses to chronic intermittent hypoxia in the frequently sampled intravenous glucose tolerance test. J. Appl. Physiol. 2017, 122, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bosch-Marce, M.; Nanayakkara, A.; Savransky, V.; Fried, S.K.; Semenza, G.L.; Polotsky, V.Y. Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia-inducible factor-1alpha. Physiol. Genom. 2006, 25, 450–457. [Google Scholar] [CrossRef]
- Drager, L.F.; Li, J.; Shin, M.-K.; Reinke, C.; Aggarwal, N.R.; Jun, J.C.; Bevans-Fonti, S.; Sztalryd, C.; O’Byrne, S.M.; Kroupa, O.; et al. Intermittent hypoxia inhibits clearance of triglyceride-rich lipoproteins and inactivates adipose lipoprotein lipase in a mouse model of sleep apnoea. Eur. Heart J. 2012, 33, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.C.; Shin, M.-K.; Yao, Q.; Bevans-Fonti, S.; Poole, J.; Drager, L.F.; Polotsky, V.Y. Acute hypoxia induces hypertriglyceridemia by decreasing plasma triglyceride clearance in mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E377–E388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drager, L.F.; Yao, Q.; Hernandez, K.L.; Shin, M.-K.; Bevans-Fonti, S.; Gay, J.; Sussan, T.E.; Jun, J.C.; Myers, A.C.; Olivecrona, G.; et al. Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin-like 4. Am. J. Respir. Crit. Care Med. 2013, 188, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q.; Shin, M.-K.; Jun, J.C.; Hernandez, K.L.; Aggarwal, N.R.; Mock, J.R.; Gay, J.; Drager, L.F.; Polotsky, V.Y. Effect of chronic intermittent hypoxia on triglyceride uptake in different tissues. J. Lipid Res. 2013, 54, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- Savransky, V.; Jun, J.; Li, J.; Nanayakkara, A.; Fonti, S.; Moser, A.B.; Steele, K.E.; Schweitzer, M.A.; Patil, S.P.; Bhanot, S.; et al. Dyslipidemia and atherosclerosis induced by chronic intermittent hypoxia are attenuated by deficiency of stearoyl coenzyme A desaturase. Circ. Res. 2008, 103, 1173–1180. [Google Scholar] [CrossRef]
- Jun, J.; Reinke, C.; Bedja, D.; Berkowitz, D.; Bevans-Fonti, S.; Li, J.; Barouch, L.A.; Gabrielson, K.; Polotsky, V.Y. Effect of intermittent hypoxia on atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 2010, 209, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Kline, D.D.; Dick, T.E.; Prabhakar, N.R. Chronic intermittent hypoxia enhances carotid body chemoreceptor response to low oxygen. Adv. Exp. Med. Biol. 2001, 499, 33–38. [Google Scholar] [CrossRef]
- Peng, Y.-J.; Prabhakar, N.R. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J. Appl. Physiol. 2004, 96, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-J.; Overholt, J.L.; Kline, D.; Kumar, G.K.; Prabhakar, N.R. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: Implications for recurrent apneas. Proc. Natl. Acad. Sci. USA 2003, 100, 10073–10078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.-J.; Makarenko, V.V.; Nanduri, J.; Vasavda, C.; Raghuraman, G.; Yuan, G.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. Inherent variations in CO-H2S-mediated carotid body O2 sensing mediate hypertension and pulmonary edema. Proc. Natl. Acad. Sci. USA 2014, 111, 1174–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, D.D.; Peng, Y.-J.; Manalo, D.J.; Semenza, G.L.; Prabhakar, N.R. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.R.; Semenza, G.L. Regulation of Carotid Body Oxygen Sensing by Hypoxia-Inducible Factors. Pflugers Arch. 2016, 468, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.-K.; Yao, Q.; Jun, J.C.; Bevans-Fonti, S.; Yoo, D.-Y.; Han, W.; Mesarwi, O.; Richardson, R.; Fu, Y.-Y.; Pasricha, P.J.; et al. Carotid body denervation prevents fasting hyperglycemia during chronic intermittent hypoxia. J. Appl. Physiol. 2014, 117, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, E.C.; Lesske, J.; Behm, R.; Miller, C.C.; Stauss, H.; Unger, T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J. Appl. Physiol. 1992, 72, 1978–1984. [Google Scholar] [CrossRef]
- McBryde, F.D.; Abdala, A.P.; Hendy, E.B.; Pijacka, W.; Marvar, P.; Moraes, D.J.A.; Sobotka, P.A.; Paton, J.F.R. The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat. Commun. 2013, 4, 2395. [Google Scholar] [CrossRef] [Green Version]
- Paton, J.F.R.; Sobotka, P.A.; Fudim, M.; Engelman, Z.J.; Engleman, Z.J.; Hart, E.C.J.; McBryde, F.D.; Abdala, A.P.; Marina, N.; Gourine, A.V.; et al. The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 2013, 61, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, H.E.; Sica, A.; Batson, D.; Scharf, S.M. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J. Appl. Physiol. 1999, 86, 298–305. [Google Scholar] [CrossRef]
- Dick, T.E.; Hsieh, Y.-H.; Wang, N.; Prabhakar, N. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp. Physiol. 2007, 92, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Zoccal, D.B.; Simms, A.E.; Bonagamba, L.G.H.; Braga, V.A.; Pickering, A.E.; Paton, J.F.R.; Machado, B.H. Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J. Physiol. 2008, 586, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lusina, S.; Xie, T.; Ji, E.; Xiang, S.; Liu, Y.; Weiss, J.W. Sympathetic response to chemostimulation in conscious rats exposed to chronic intermittent hypoxia. Respir. Physiol. Neurobiol. 2009, 166, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.; Poucher, S.M.; Summerill, R.A. The effects of stimulating carotid chemoreceptors on renal haemodynamics and function in dogs. J. Physiol. 1987, 392, 451–462. [Google Scholar] [CrossRef]
- Prabhakar, N.R. Carotid body chemoreflex: A driver of autonomic abnormalities in sleep apnoea. Exp. Physiol. 2016, 101, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Hedner, J.A.; Wilcox, I.; Laks, L.; Grunstein, R.R.; Sullivan, C.E. A specific and potent pressor effect of hypoxia in patients with sleep apnea. Am. Rev. Respir. Dis. 1992, 146, 1240–1245. [Google Scholar] [CrossRef]
- Gerst, D.G.; Yokhana, S.S.; Carney, L.M.; Lee, D.S.; Badr, M.S.; Qureshi, T.; Anthouard, M.N.; Mateika, J.H. The hypoxic ventilatory response and ventilatory long-term facilitation are altered by time of day and repeated daily exposure to intermittent hypoxia. J. Appl. Physiol. 2011, 110, 15–28. [Google Scholar] [CrossRef]
- Imadojemu, V.A.; Mawji, Z.; Kunselman, A.; Gray, K.S.; Hogeman, C.S.; Leuenberger, U.A. Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest 2007, 131, 1406–1413. [Google Scholar] [CrossRef]
- Trombetta, I.C.; Maki-Nunes, C.; Toschi-Dias, E.; Alves, M.-J.N.N.; Rondon, M.U.P.B.; Cepeda, F.X.; Drager, L.F.; Braga, A.M.F.W.; Lorenzi-Filho, G.; Negrao, C.E. Obstructive sleep apnea is associated with increased chemoreflex sensitivity in patients with metabolic syndrome. Sleep 2013, 36, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Spicuzza, L.; Bernardi, L.; Balsamo, R.; Ciancio, N.; Polosa, R.; Di Maria, G. Effect of treatment with nasal continuous positive airway pressure on ventilatory response to hypoxia and hypercapnia in patients with sleep apnea syndrome. Chest 2006, 130, 774–779. [Google Scholar] [CrossRef]
- Reeves, S.R.; Gozal, E.; Guo, S.Z.; Sachleben, L.R.; Brittian, K.R.; Lipton, A.J.; Gozal, D. Effect of long-term intermittent and sustained hypoxia on hypoxic ventilatory and metabolic responses in the adult rat. J. Appl. Physiol. 2003, 95, 1767–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, D.D.; Ramirez-Navarro, A.; Kunze, D.L. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: Evidence for homeostatic plasticity. J. Neurosci. 2007, 27, 4663–4673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Silva, J.H.; Zoccal, D.B.; Machado, B.H. Chronic intermittent hypoxia alters glutamatergic control of sympathetic and respiratory activities in the commissural NTS of rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R785–R793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haselton, J.R.; Guyenet, P.G. Central respiratory modulation of medullary sympathoexcitatory neurons in rat. Am. J. Physiol. 1989, 256, R739–R750. [Google Scholar] [CrossRef] [PubMed]
- Moraes, D.J.A.; da Silva, M.P.; Bonagamba, L.G.H.; Mecawi, A.S.; Zoccal, D.B.; Antunes-Rodrigues, J.; Varanda, W.A.; Machado, B.H. Electrophysiological properties of rostral ventrolateral medulla presympathetic neurons modulated by the respiratory network in rats. J. Neurosci. 2013, 33, 19223–19237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, G.K.; Rai, V.; Sharma, S.D.; Ramakrishnan, D.P.; Peng, Y.-J.; Souvannakitti, D.; Prabhakar, N.R. Chronic intermittent hypoxia induces hypoxia-evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J. Physiol. 2006, 575, 229–239. [Google Scholar] [CrossRef]
- Hui, A.S.; Striet, J.B.; Gudelsky, G.; Soukhova, G.K.; Gozal, E.; Beitner-Johnson, D.; Guo, S.-Z.; Sachleben, L.R.; Haycock, J.W.; Gozal, D.; et al. Regulation of Catecholamines by Sustained and Intermittent Hypoxia in Neuroendocrine Cells and Sympathetic Neurons. Hypertension 2003, 42, 1130–1136. [Google Scholar] [CrossRef] [Green Version]
- Paton, J.F.R.; Ratcliffe, L.; Hering, D.; Wolf, J.; Sobotka, P.A.; Narkiewicz, K. Revelations About Carotid Body Function Through its Pathological Role in Resistant Hypertension. Curr. Hypertens. Rep. 2013, 15, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.M.; Metcalfe, J.D. Cardiovascular changes associated with augmented breaths in normoxia and hypoxia in the rat. J. Physiol. 1988, 400, 15–27. [Google Scholar] [CrossRef] [Green Version]
- De Burgh Daly, M.; Wood, L.M.; Ward, J. Cardiovascular responses to carotid chemoreceptor stimulation in the dog: Their modulation by urinary bladder distension. J. Physiol. 2000, 524, 903–917. [Google Scholar] [CrossRef]
- Tan, J.; Xiong, B.; Zhu, Y.; Yao, Y.; Qian, J.; Rong, S.; Yang, G.; Zhu, Q.; Jiang, Y.; Zhou, Q.; et al. Carotid body enlargement in hypertension and other comorbidities evaluated by ultrasonography. J. Hypertens. 2019, 37, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Honig, A.; Habeck, J.O.; Pfeiffer, C.; Schmidt, M.; Huckstorf, C.; Rotter, H.; Eckermann, P. The carotid bodies of spontaneously hypertensive rats (SHR): A functional and morphologic study. Acta Biol. Med. Ger. 1981, 40, 1021–1030. [Google Scholar] [PubMed]
- Clarke, J.A.; de Daly, M.B.; Ead, H.W. Vascular Analysis of the Carotid Body in the Spontaneously Hypertensive Rat. In Neurobiology and Cell Physiology of Chemoreception; Data, P.G., Acker, H., Lahiri, S., Eds.; Springer Science & Business Media: Boston, MA, USA, 1993; pp. 3–8. ISBN 978-1-4615-2966-8. [Google Scholar]
- Takahashi, M.; Matsuda, H.; Hayashida, Y.; Yamamoto, Y.; Tsukuda, M.; Kusakabe, T. Morphological characteristics and peptidergic innervation in the carotid body of spontaneously hypertensive rats. Histol. Histopathol. 2011, 26, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Somers, V.K.; Mark, A.L.; Abboud, F.M. Potentiation of sympathetic nerve responses to hypoxia in borderline hypertensive subjects. Hypertension 1988, 11, 608–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, M.R.; Forster, H.V.; Papanek, P.E.; Dwinell, M.R.; Hogan, G.E. Ventilatory phenotypes among four strains of adult rats. J. Appl. Physiol. 2002, 93, 974–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Z.-Y.; Lu, Y.; Whiteis, C.A.; Simms, A.E.; Paton, J.F.R.; Chapleau, M.W.; Abboud, F.M. Chemoreceptor hypersensitivity, sympathetic excitation and overexpression of ASIC and TASK channels prior to hypertension in SHR. Circ. Res. 2010, 106, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Hayward, L.F.; Castellanos, M.; Noah, C. Cardiorespiratory variability following repeat acute hypoxia in the conscious SHR versus two normotensive rat strains. Auton. Neurosci. 2012, 171, 58–65. [Google Scholar] [CrossRef]
- Simms, A.E.; Paton, J.F.R.; Pickering, A.E.; Allen, A.M. Amplified respiratory–sympathetic coupling in the spontaneously hypertensive rat: Does it contribute to hypertension? J. Physiol. 2009, 587, 597–610. [Google Scholar] [CrossRef]
- Siński, M.; Lewandowski, J.; Przybylski, J.; Bidiuk, J.; Abramczyk, P.; Ciarka, A.; Gaciong, Z. Tonic activity of carotid body chemoreceptors contributes to the increased sympathetic drive in essential hypertension. Hypertens. Res. 2012, 35, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.A.J.; Timmers, H.J.L.M.; Wieling, W.; Wagenaar, M.; Marres, H.A.M.; Lenders, J.W.M.; van Montfrans, G.A.; Karemaker, J.M. Long-term effects of carotid sinus denervation on arterial blood pressure in humans. Circulation 2002, 105, 1329–1335. [Google Scholar] [CrossRef] [Green Version]
- Fudim, M.; Groom, K.L.; Laffer, C.L.; Netterville, J.L.; Robertson, D.; Elijovich, F. Effects of Carotid Body Tumor Resection on the Blood Pressure of Essential Hypertensive Patients. J. Am. Soc. Hypertens. 2015, 9, 435–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapp, S.; Aller, M.I.; Wisden, W.; Gourine, A.V. A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J. Neurosci. 2008, 28, 8844–8850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Sáenz, P.; Levitsky, K.L.; Marcos-Almaraz, M.T.; Bonilla-Henao, V.; Pascual, A.; López-Barneo, J. Carotid body chemosensory responses in mice deficient of TASK channels. J. Gen. Physiol. 2010, 135, 379–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulkey, D.K.; Talley, E.M.; Stornetta, R.L.; Siegel, A.R.; West, G.H.; Chen, X.; Sen, N.; Mistry, A.M.; Guyenet, P.G.; Bayliss, D.A. TASK channels determine pH sensitivity in select respiratory neurons but do not contribute to central respiratory chemosensitivity. J. Neurosci. 2007, 27, 14049–14058. [Google Scholar] [CrossRef]
- Shin, M.-K.; Eraso, C.C.; Mu, Y.-P.; Gu, C.; Yeung, B.H.Y.; Kim, L.J.; Liu, X.-R.; Wu, Z.-J.; Paudel, O.; Pichard, L.E.; et al. Leptin Induces Hypertension Acting on Transient Receptor Potential Melastatin 7 Channel in the Carotid Body. Circ. Res. 2019, 125, 989–1002. [Google Scholar] [CrossRef]
- Shirahata, M.; Tang, W.-Y.; Shin, M.-K.; Polotsky, V.Y. Is the Carotid Body a Metabolic Monitor? Adv. Exp. Med. Biol. 2015, 860, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Caballero-Eraso, C.; Shin, M.-K.; Pho, H.; Kim, L.J.; Pichard, L.E.; Wu, Z.-J.; Gu, C.; Berger, S.; Pham, L.; Yeung, H.-Y.B.; et al. Leptin acts in the carotid bodies to increase minute ventilation during wakefulness and sleep and augment the hypoxic ventilatory response. J. Physiol. 2019, 597, 151–172. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Prabhakar, N.R. Neural regulation of hypoxia-inducible factors and redox state drives the pathogenesis of hypertension in a rodent model of sleep apnea. J. Appl. Physiol. 2015, 119, 1152–1156. [Google Scholar] [CrossRef] [Green Version]
- Schultz, H.D. Angiotensin and Carotid Body Chemoreception in Heart Failure. Curr. Opin. Pharmacol. 2011, 11, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Li, Y.-L.; Schultz, H.D. Role of blood flow in carotid body chemoreflex function in heart failure. J. Physiol. 2011, 589, 245–258. [Google Scholar] [CrossRef]
- Clarke, J.A.; Daly, M.D.B.; Ead, H.W.; Hennessy, E.M. The carotid body of the spontaneous insulin-dependent diabetic rat. Braz. J. Med. Biol. Res. 1999, 32, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, E.; Sacramento, J.F.; Melo, B.F.; Conde, S.V. Carotid Body Dysfunction in Diet-Induced Insulin Resistance Is Associated with Alterations in Its Morphology. Adv. Exp. Med. Biol. 2018, 1071, 103–108. [Google Scholar] [CrossRef]
- Alvarez-Buylla, R.; de Alvarez-Buylla, E.R. Carotid sinus receptors participate in glucose homeostasis. Respir. Physiol. 1988, 72, 347–359. [Google Scholar] [CrossRef]
- Pardal, R.; López-Barneo, J. Low glucose–sensing cells in the carotid body. Nat. Neurosci. 2002, 5, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Coker, R.H.; Stone, E.E.; Lacy, D.B.; Jabbour, K.; Williams, P.E.; Wasserman, D.H. Evidence that carotid bodies play an important role in glucoregulation in vivo. Diabetes 2000, 49, 1434–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, D.S.; Voter, W.A.; Karan, S. The effects of hypo- and hyperglycaemia on the hypoxic ventilatory response in humans. J. Physiol. 2007, 582, 859–869. [Google Scholar] [CrossRef]
- Wehrwein, E.A.; Basu, R.; Basu, A.; Curry, T.B.; Rizza, R.A.; Joyner, M.J. Hyperoxia blunts counterregulation during hypoglycaemia in humans: Possible role for the carotid bodies? J. Physiol. 2010, 588, 4593–4601. [Google Scholar] [CrossRef]
- Limberg, J.K.; Taylor, J.L.; Dube, S.; Basu, R.; Basu, A.; Joyner, M.J.; Wehrwein, E.A. Role of the carotid body chemoreceptors in baroreflex control of blood pressure during hypoglycaemia in humans. Exp. Physiol. 2014, 99, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Buttigieg, J.; Nurse, C.A. Neurotransmitter mechanisms mediating low-glucose signalling in cocultures and fresh tissue slices of rat carotid body. J. Physiol. 2007, 578, 735–750. [Google Scholar] [CrossRef]
- Bin-Jaliah, I.; Maskell, P.D.; Kumar, P. Indirect sensing of insulin-induced hypoglycaemia by the carotid body in the rat. J. Physiol. 2004, 556, 255–266. [Google Scholar] [CrossRef]
- Conde, S.V.; Obeso, A.; Gonzalez, C. Low glucose effects on rat carotid body chemoreceptor cells’ secretory responses and action potential frequency in the carotid sinus nerve. J. Physiol. 2007, 585, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Martin, T.; Fernandez-Martinez, S.; Rigual, R.; Obeso, A.; Gonzalez, C. Effects of low glucose on carotid body chemoreceptor cell activity studied in cultures of intact organs and in dissociated cells. Am. J. Physiol. Cell Physiol. 2012, 302, C1128–C1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, A.P.; Turner, P.J.; Carter, P.; Leadbeater, W.; Ray, C.J.; Hauton, D.; Buckler, K.J.; Kumar, P. Glycogen metabolism protects against metabolic insult to preserve carotid body function during glucose deprivation. J. Physiol. 2014, 592, 4493–4506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakhar, N.R.; Joyner, M.J. Tasting arterial blood: What do the carotid chemoreceptors sense? Front. Physiol. 2015, 5, 524. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.L.; Ray, C.J.; Holmes, A.P.; Pye, R.L.; Wyatt, C.N.; Coney, A.M.; Kumar, P. Adrenaline release evokes hyperpnoea and an increase in ventilatory CO2 sensitivity during hypoglycaemia: A role for the carotid body. J. Physiol. 2016, 594, 4439–4452. [Google Scholar] [CrossRef] [Green Version]
- García-Fernández, M.; Ortega-Sáenz, P.; Castellano, A.; López-Barneo, J. Mechanisms of low-glucose sensitivity in carotid body glomus cells. Diabetes 2007, 56, 2893–2900. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Ortega-Sáenz, P.; García-Fernández, M.; González-Rodríguez, P.; Caballero-Eraso, C.; López-Barneo, J. Glucose sensing by carotid body glomus cells: Potential implications in disease. Front. Physiol. 2014, 5, 398. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.J.; Ortega, F.E.; Riegler, J.; Madison, D.V.; Krasnow, M.A. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 2015, 527, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-J.; Gridina, A.; Nanduri, J.; Fox, A.P.; Prabhakar, N.R. Impaired carotid body hypoxic sensing in mice deficient in olfactory receptor 78. bioRxiv 2019, 757120. [Google Scholar] [CrossRef]
- Taylor, S.C.; Shaw, S.M.; Peers, C. Mitochondrial inhibitors evoke catecholamine release from pheochromocytoma cells. Biochem. Biophys. Res. Commun. 2000, 273, 17–21. [Google Scholar] [CrossRef]
- Ortega-Sáenz, P.; Pardal, R.; García-Fernandez, M.; López-Barneo, J. Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J. Physiol. 2003, 548, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Agüera, M.C.; Gao, L.; González-Rodríguez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Gaggini, M.; DeFronzo, R.A. Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results from the San Antonio Metabolism Study. Diabetes 2017, 66, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Hædersdal, S.; Lund, A.; Knop, F.K.; Vilsbøll, T. The Role of Glucagon in the Pathophysiology and Treatment of Type 2 Diabetes. Mayo Clin. Proc. 2018, 93, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Coker, R.H.; Denny, J.C.; Lacy, D.B.; Jabbour, K.; Williams, P.E.; Wasserman, D.H. Role of carotid bodies in control of the neuroendocrine response to exercise. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E742–E748. [Google Scholar] [CrossRef]
- Johnson, B.D.; Peinado, A.B.; Ranadive, S.M.; Curry, T.B.; Joyner, M.J. Effects of intravenous low-dose dopamine infusion on glucose regulation during prolonged aerobic exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R49–R57. [Google Scholar] [CrossRef]
- Newhouse, L.P.; Joyner, M.J.; Curry, T.B.; Laurenti, M.C.; Man, C.D.; Cobelli, C.; Vella, A.; Limberg, J.K. Three hours of intermittent hypoxia increases circulating glucose levels in healthy adults. Physiol. Rep. 2017, 5, e13106. [Google Scholar] [CrossRef]
- Ribeiro, M.J.; Sacramento, J.F.; Gonzalez, C.; Guarino, M.P.; Monteiro, E.C.; Conde, S.V. Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes 2013, 62, 2905–2916. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.A.; Hoffman, R.P.; Balon, T.W.; Sinkey, C.A.; Mark, A.L. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J. Clin. Investig. 1991, 87, 2246–2252. [Google Scholar] [CrossRef]
- Tomiyama, H.; Kushiro, T.; Abeta, H.; Kurumatani, H.; Taguchi, H.; Kuga, N.; Saito, F.; Kobayashi, F.; Otsuka, Y.; Kanmatsuse, K. Blood pressure response to hyperinsulinemia in salt-sensitive and salt-resistant rats. Hypertension 1992, 20, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, G.W.; Straznicky, N.E.; Lambert, E.A.; Dixon, J.B.; Schlaich, M.P. Sympathetic nervous activation in obesity and the metabolic syndrome—Causes, consequences and therapeutic implications. Pharmacol. Therapeut. 2010, 126, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Pereda, S.A.; Eckstein, J.W.; Abboud, F.M. Cardiovascular responses to insulin in the absence of hypoglycemia. Am. J. Physiol. Leg. Content 1962, 202, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Sacramento, J.F.; Ribeiro, M.J.; Rodrigues, T.; Olea, E.; Melo, B.F.; Guarino, M.P.; Fonseca-Pinto, R.; Ferreira, C.R.; Coelho, J.; Obeso, A.; et al. Functional abolition of carotid body activity restores insulin action and glucose homeostasis in rats: Key roles for visceral adipose tissue and the liver. Diabetologia 2017, 60, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limberg, J.K.; Mozer, M.T.; Holbein, W.W.; Johnson, B.D.; Prabhakar, N.R.; Curry, T.B.; Joyner, M.J. Low-dose Dopamine, but Not Acute Hyperoxia, Attenuates the Sympathoexcitatory Response to Hyperinsulinemia in Healthy Humans. FASEB J. 2017, 31, 867.4. [Google Scholar] [CrossRef]
- Limberg, J.K. Glucose, insulin, and the carotid body chemoreceptors in humans. Physiol. Genom. 2018, 50, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Joyner, M.J.; Limberg, J.K.; Wehrwein, E.A.; Johnson, B.D. Role of the carotid body chemoreceptors in glucose homeostasis and thermoregulation in humans. J. Physiol. 2018, 596, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.H.; Pessin, J.E. Insulin regulation of glucose uptake: A complex interplay of intracellular signalling pathways. Diabetologia 2002, 45, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Freychet, L.; Rizkalla, S.W.; Desplanque, N.; Basdevant, A.; Zirinis, P.; Tchobroutsky, G.; Slama, G. Effect of intranasal glucagon on blood glucose levels in healthy subjects and hypoglycaemic patients with insulin-dependent diabetes. Lancet 1988, 331, 1364–1366. [Google Scholar] [CrossRef]
- Cunha-Guimaraes, J.P.; Guarino, M.P.; Timóteo, A.T.; Caires, I.; Sacramento, J.F.; Ribeiro, M.J.; Selas, M.; Santiago, J.C.P.; Mota-Carmo, M.; Conde, S.V. Carotid body chemosensitivity: Early biomarker of dysmetabolism in humans. Eur. J. Endocrinol. 2020, 182, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Vera-Cruz, P.; Guerreiro, F.; Ribeiro, M.J.; Guarino, M.P.; Conde, S.V. Hyperbaric Oxygen Therapy Improves Glucose Homeostasis in Type 2 Diabetes Patients: A Likely Involvement of the Carotid Bodies. Adv. Exp. Med. Biol. 2015, 860, 221–225. [Google Scholar] [CrossRef]
- Cracchiolo, M.; Sacramento, J.F.; Mazzoni, A.; Panarese, A.; Carpaneto, J.; Conde, S.V.; Micera, S. Decoding Neural Metabolic Markers From the Carotid Sinus Nerve in a Type 2 Diabetes Model. IEEE Trans. Neural Syst. Rehabil. Eng. 2019, 27, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.R.; Himal, H.S.; Rebuck, A.S. Ventilatory responses to hypercapnia and hypoxia in patients with eucapnic morbid obesity before and after weight loss. Clin. Sci. 1990, 78, 541–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narkiewicz, K.; Kato, M.; Pesek, C.A.; Somers, V.K. Human Obesity Is Characterized by a Selective Potentiation of Central Chemoreflex Sensitivity. Hypertension 1999, 33, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Piper, A.J.; Grunstein, R.R. Obesity Hypoventilation Syndrome. Am. J. Respir. Crit. Care Med. 2011, 183, 292–298. [Google Scholar] [CrossRef]
- Strohl, K.P.; Thomas, A.J.; Jean, P.S.; Schlenker, E.H.; Koletsky, R.J.; Schork, N.J. Ventilation and metabolism among rat strains. J. Appl. Physiol. 1997, 82, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Zwillich, C.W.; Sutton, F.D.; Pierson, D.J.; Creagh, E.M.; Weil, J.V. Decreased hypoxic ventilatory drive in the obesity-hypoventilation syndrome. Am. J. Med. 1975, 59, 343–348. [Google Scholar] [CrossRef]
- Jokic, R.; Zintel, T.; Sridhar, G.; Gallagher, C.G.; Fitzpatrick, M.F. Ventilatory responses to hypercapnia and hypoxia in relatives of patients with the obesity hypoventilation syndrome. Thorax 2000, 55, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Haslam, D.W.; James, W.P.T. Obesity. Lancet 2005, 366, 1197–1209. [Google Scholar] [CrossRef]
- Schwartz, A.R.; Patil, S.P.; Laffan, A.M.; Polotsky, V.; Schneider, H.; Smith, P.L. Obesity and obstructive sleep apnea: Pathogenic mechanisms and therapeutic approaches. Proc. Am. Thorac. Soc. 2008, 5, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Flier, J.S. Obesity and the regulation of energy balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M. Leptin at 14 y of age: An ongoing story1234. Am. J. Clin. Nutr. 2009, 89, 973S–979S. [Google Scholar] [CrossRef] [Green Version]
- O’donnell, C.P.; Schaub, C.D.; Haines, A.S.; Berkowitz, D.E.; Tankersley, C.G.; Schwartz, A.R.; Smith, P.L. Leptin prevents respiratory depression in obesity. Am. J. Respir. Crit. Care Med. 1999, 159, 1477–1484. [Google Scholar] [CrossRef]
- Inyushkina, E.M.; Merkulova, N.A.; Inyushkin, A.N. Mechanisms of the respiratory activity of leptin at the level of the solitary tract nucleus. Neurosci. Behav. Physiol. 2010, 40, 707–713. [Google Scholar] [CrossRef]
- Bassi, M.; Furuya, W.I.; Menani, J.V.; Colombari, D.S.A.; do Carmo, J.M.; da Silva, A.A.; Hall, J.E.; Moreira, T.S.; Wenker, I.C.; Mulkey, D.K.; et al. Leptin into the ventrolateral medulla facilitates chemorespiratory response in leptin-deficient (ob/ob) mice. Acta Physiol. 2014, 211, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Pho, H.; Kirkness, J.; Ladenheim, E.E.; Bi, S.; Moran, T.H.; Fuller, D.D.; Schwartz, A.R.; Polotsky, V.Y. Localizing Effects of Leptin on Upper Airway and Respiratory Control during Sleep. Sleep 2016, 39, 1097–1106. [Google Scholar] [CrossRef]
- Banks, W.A.; DiPalma, C.R.; Farrell, C.L. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides 1999, 20, 1341–1345. [Google Scholar] [CrossRef]
- Wauman, J.; Tavernier, J. Leptin receptor signaling: Pathways to leptin resistance. Front. Biosci. 2011, 16, 2771–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, B.G.; Kato, M.; Narkiewicz, K.; Choe, I.; Somers, V.K. Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H234–H237. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Patterson, M.; Twigg, G.L.; Vazir, A.; Ghatei, M.; Morrell, M.J.; Polkey, M.I. Lack of association between impaired glucose tolerance and appetite regulating hormones in patients with obstructive sleep apnea. J. Clin. Sleep Med. 2011, 7, 486B–492B. [Google Scholar] [CrossRef] [Green Version]
- Gaines, J.; Vgontzas, A.N.; Fernandez-Mendoza, J.; Calhoun, S.L.; He, F.; Liao, D.; Sawyer, M.D.; Bixler, E.O. Inflammation mediates the association between visceral adiposity and obstructive sleep apnea in adolescents. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E851–E858. [Google Scholar] [CrossRef]
- Hirotsu, C.; Albuquerque, R.G.; Nogueira, H.; Hachul, H.; Bittencourt, L.; Tufik, S.; Andersen, M.L. The relationship between sleep apnea, metabolic dysfunction and inflammation: The gender influence. Brain Behav. Immun. 2017, 59, 211–218. [Google Scholar] [CrossRef]
- Imayama, I.; Prasad, B. Role of Leptin in Obstructive Sleep Apnea. Ann. Am. Thorac. Soc. 2017, 14, 1607–1621. [Google Scholar] [CrossRef]
- Ip, M.S.; Lam, K.S.; Ho, C.; Tsang, K.W.; Lam, W. Serum leptin and vascular risk factors in obstructive sleep apnea. Chest 2000, 118, 580–586. [Google Scholar] [CrossRef]
- Kapsimalis, F.; Varouchakis, G.; Manousaki, A.; Daskas, S.; Nikita, D.; Kryger, M.; Gourgoulianis, K. Association of sleep apnea severity and obesity with insulin resistance, C-reactive protein, and leptin levels in male patients with obstructive sleep apnea. Lung 2008, 186, 209–217. [Google Scholar] [CrossRef]
- Schwab, R.J.; Pasirstein, M.; Pierson, R.; Mackley, A.; Hachadoorian, R.; Arens, R.; Maislin, G.; Pack, A.I. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am. J. Respir. Crit. Care Med. 2003, 168, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.B.; Kirkness, J.P.; Smith, P.L.; Schneider, H.; Polotsky, M.; Richardson, R.A.; Hernandez, W.C.; Schwartz, A.R. Novel whole body plethysmography system for the continuous characterization of sleep and breathing in a mouse. J. Appl. Physiol. 2012, 112, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pho, H.; Hernandez, A.B.; Arias, R.S.; Leitner, E.B.; Van Kooten, S.; Kirkness, J.P.; Schneider, H.; Smith, P.L.; Polotsky, V.Y.; Schwartz, A.R. The effect of leptin replacement on sleep-disordered breathing in the leptin-deficient ob/ob mouse. J. Appl. Physiol. 2016, 120, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleury Curado, T.; Pho, H.; Berger, S.; Caballero-Eraso, C.; Shin, M.-K.; Sennes, L.U.; Pham, L.; Schwartz, A.R.; Polotsky, V.Y. Sleep-disordered breathing in C57BL/6J mice with diet-induced obesity. Sleep 2018, 41, zsy089. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Pho, H.; Fleury-Curado, T.; Bevans-Fonti, S.; Younas, H.; Shin, M.-K.; Jun, J.C.; Anokye-Danso, F.; Ahima, R.S.; Enquist, L.W.; et al. Intranasal Leptin Relieves Sleep-disordered Breathing in Mice with Diet-induced Obesity. Am. J. Respir. Crit. Care Med. 2019, 199, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Porzionato, A.; Rucinski, M.; Macchi, V.; Stecco, C.; Castagliuolo, I.; Malendowicz, L.K.; De Caro, R. Expression of leptin and leptin receptor isoforms in the rat and human carotid body. Brain Res. 2011, 1385, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Messenger, S.A.; Moreau, J.M.; Ciriello, J. Intermittent hypoxia and systemic leptin administration induces pSTAT3 and Fos/Fra-1 in the carotid body. Brain Res. 2012, 1446, 56–70. [Google Scholar] [CrossRef]
- Messenger, S.A.; Ciriello, J. Effects of intermittent hypoxia on leptin signalling in the carotid body. Neuroscience 2013, 232, 216–225. [Google Scholar] [CrossRef]
- Yuan, F.; Wang, H.; Feng, J.; Wei, Z.; Yu, H.; Zhang, X.; Zhang, Y.; Wang, S. Leptin Signaling in the Carotid Body Regulates a Hypoxic Ventilatory Response Through Altering TASK Channel Expression. Front. Physiol. 2018, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.J.; Sacramento, J.F.; Gallego-Martin, T.; Olea, E.; Melo, B.F.; Guarino, M.P.; Yubero, S.; Obeso, A.; Conde, S.V. High fat diet blunts the effects of leptin on ventilation and on carotid body activity. J. Physiol. 2018, 596, 3187–3199. [Google Scholar] [CrossRef]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef]
- Kelesidis, T.; Kelesidis, I.; Chou, S.; Mantzoros, C.S. Narrative review: The role of leptin in human physiology: Emerging clinical applications. Ann. Intern. Med. 2010, 152, 93–100. [Google Scholar] [CrossRef]
- Ciriello, J.; Moreau, J.M. Systemic administration of leptin potentiates the response of neurons in the nucleus of the solitary tract to chemoreceptor activation in the rat. Neuroscience 2013, 229, 88–99. [Google Scholar] [CrossRef]
- Porzionato, A.; Macchi, V.; De Caro, R. Role of the Carotid Body in Obesity-Related Sympathoactivation. Hypertension 2013, 61, e57. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, R.; Andrade, D.C.; Lucero, C.; Arias, P.; Iturriaga, R. Carotid Body Ablation Abrogates Hypertension and Autonomic Alterations Induced by Intermittent Hypoxia in Rats. Hypertension 2016, 68, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Rabl, H.; Friehs, I.; Gutschi, S.; Pascher, O.; Koch, G. Diagnosis and treatment of carotid body tumors. Thorac. Cardiovasc. Surg. 1993, 41, 340–343. [Google Scholar] [CrossRef]
- Narkiewicz, K.; Ratcliffe, L.E.K.; Hart, E.C.; Briant, L.J.B.; Chrostowska, M.; Wolf, J.; Szyndler, A.; Hering, D.; Abdala, A.P.; Manghat, N.; et al. Unilateral Carotid Body Resection in Resistant Hypertension. JACC. Basic Transl. Sci. 2016, 1, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Wehrwein, E.A.; Limberg, J.K.; Taylor, J.L.; Dube, S.; Basu, A.; Basu, R.; Rizza, R.A.; Curry, T.B.; Joyner, M.J. Effect of bilateral carotid body resection on the counterregulatory response to hypoglycaemia in humans. Exp. Physiol. 2015, 100, 69–78. [Google Scholar] [CrossRef]
- Powell, F.L. The influence of chronic hypoxia upon chemoreception. Respir. Physiol. Neurobiol. 2007, 157, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, H.V. Plasticity in the control of breathing following sensory denervation. J. Appl. Physiol. 2003, 94, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.B.; Vidruk, E.H.; Dempsey, J.A. Carotid body excision significantly changes ventilatory control in awake rats. J. Appl. Physiol. 1988, 64, 666–671. [Google Scholar] [CrossRef]
- Dahan, A.; Nieuwenhuijs, D.; Teppema, L. Plasticity of Central Chemoreceptors: Effect of Bilateral Carotid Body Resection on Central CO2 Sensitivity. PLoS Med. 2007, 4, e239. [Google Scholar] [CrossRef] [Green Version]
- Limberg, J.K.; Taylor, J.L.; Mozer, M.T.; Dube, S.; Basu, A.; Basu, R.; Rizza, R.A.; Curry, T.B.; Joyner, M.J.; Wehrwein, E.A. Effect of Bilateral Carotid Body Resection on Cardiac Baroreflex Control of Blood Pressure During Hypoglycemia. Hypertension 2015, 65, 1365–1371. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Prabhakar, N.R. The role of hypoxia-inducible factors in carotid body (patho) physiology. J. Physiol. 2018, 596, 2977–2983. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.R.; Peng, Y.-J.; Yuan, G.; Nanduri, J. Reactive oxygen radicals and gaseous transmitters in carotid body activation by intermittent hypoxia. Cell Tissue Res. 2018, 372, 427–431. [Google Scholar] [CrossRef]
- Yuan, G.; Adhikary, G.; McCormick, A.A.; Holcroft, J.J.; Kumar, G.K.; Prabhakar, N.R. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J. Physiol. 2004, 557, 773–783. [Google Scholar] [CrossRef]
- Peng, Y.-J.; Nanduri, J.; Yuan, G.; Wang, N.; Deneris, E.; Pendyala, S.; Natarajan, V.; Kumar, G.K.; Prabhakar, N.R. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J. Neurosci. 2009, 29, 4903–4910. [Google Scholar] [CrossRef]
- Nanduri, J.; Wang, N.; Yuan, G.; Khan, S.A.; Souvannakitti, D.; Peng, Y.-J.; Kumar, G.K.; Garcia, J.A.; Prabhakar, N.R. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: Implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. USA 2009, 106, 1199–1204. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-J.; Yuan, G.; Khan, S.; Nanduri, J.; Makarenko, V.V.; Reddy, V.D.; Vasavda, C.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Regulation of hypoxia-inducible factor-α isoforms and redox state by carotid body neural activity in rats. J. Physiol. 2014, 592, 3841–3858. [Google Scholar] [CrossRef]
- Yuan, G.; Nanduri, J.; Khan, S.; Semenza, G.L.; Prabhakar, N.R. Induction of HIF-1alpha expression by intermittent hypoxia: Involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J. Cell. Physiol. 2008, 217, 674–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.-J.; Nanduri, J.; Khan, S.A.; Yuan, G.; Wang, N.; Kinsman, B.; Vaddi, D.R.; Kumar, G.K.; Garcia, J.A.; Semenza, G.L.; et al. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc. Natl. Acad. Sci. USA 2011, 108, 3065–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.-J.; Yuan, G.; Ramakrishnan, D.; Sharma, S.D.; Bosch-Marce, M.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Heterozygous HIF-1α deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J. Physiol. 2006, 577, 705–716. [Google Scholar] [CrossRef]
- Yuan, G.; Peng, Y.-J.; Reddy, V.D.; Makarenko, V.V.; Nanduri, J.; Khan, S.A.; Garcia, J.A.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Mutual antagonism between hypoxia-inducible factors 1α and 2α regulates oxygen sensing and cardio-respiratory homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, E1788–E1796. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-J.; Nanduri, J.; Raghuraman, G.; Souvannakitti, D.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. USA 2010, 107, 10719–10724. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.R.; Dinerman, J.L.; Agani, F.H.; Snyder, S.H. Carbon monoxide: A role in carotid body chemoreception. Proc. Natl. Acad. Sci. USA 1995, 92, 1994–1997. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-J.; Zhang, X.; Gridina, A.; Chupikova, I.; McCormick, D.L.; Thomas, R.J.; Scammell, T.E.; Kim, G.; Vasavda, C.; Nanduri, J.; et al. Complementary roles of gasotransmitters CO and H2S in sleep apnea. Proc. Natl. Acad. Sci. USA 2017, 114, 1413–1418. [Google Scholar] [CrossRef] [Green Version]
- Yuan, G.; Peng, Y.-J.; Khan, S.A.; Nanduri, J.; Singh, A.; Vasavda, C.; Semenza, G.L.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. H2S production by reactive oxygen species in the carotid body triggers hypertension in a rodent model of sleep apnea. Sci. Signal. 2016, 9, ra80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhong, H.; Vollmer, C.; Nurse, C.A. Co-release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J. Physiol. 2000, 525, 143–158. [Google Scholar] [CrossRef]
- Rong, W.; Gourine, A.V.; Cockayne, D.A.; Xiang, Z.; Ford, A.P.D.W.; Spyer, K.M.; Burnstock, G. Pivotal Role of Nucleotide P2X2 Receptor Subunit of the ATP-Gated Ion Channel Mediating Ventilatory Responses to Hypoxia. J. Neurosci. 2003, 23, 11315–11321. [Google Scholar] [CrossRef] [Green Version]
- Moraes, D.J.A.; da Silva, M.P.; Spiller, P.F.; Machado, B.H.; Paton, J.F.R. Purinergic plasticity within petrosal neurons in hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R963–R971. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.; Fearon, I.M.; Zhang, M.; Laing, M.; Vollmer, C.; Nurse, C.A. Expression of P2X2 and P2X3 receptor subunits in rat carotid body afferent neurones: Role in chemosensory signalling. J. Physiol. 2001, 537, 667–677. [Google Scholar] [CrossRef]
- Pijacka, W.; Moraes, D.J.A.; Ratcliffe, L.E.K.; Nightingale, A.K.; Hart, E.C.; da Silva, M.P.; Machado, B.H.; McBryde, F.D.; Abdala, A.P.; Ford, A.P.; et al. Purinergic receptors in the carotid body as a novel target for controlling hypertension. Nat. Med. 2016, 22, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Katayama, P.L.; Abdala, A.P.; Charles, I.; Pijacka, W.; Salgado, H.C.; Gever, J.; Ford, A.P.; Paton, J.F.R. P2X3 receptor antagonism reduces the occurrence of apnoeas in newborn rats. Respir. Physiol. Neurobiol. 2020, 277, 103438. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Sinha, A.; Ratia, K.; Yin, L.; Delgado-Rivera, L.; Petukhov, P.A.; Thatcher, G.R.J.; Wardrop, D.J. 2-Arylidene Hydrazinecarbodithioates as Potent, Selective Inhibitors of Cystathionine γ-Lyase (CSE). ACS Med. Chem. Lett. 2017, 8, 1241–1245. [Google Scholar] [CrossRef]
- Abdulqawi, R.; Dockry, R.; Holt, K.; Layton, G.; McCarthy, B.G.; Ford, A.P.; Smith, J.A. P2X3 receptor antagonist (AF-219) in refractory chronic cough: A randomised, double-blind, placebo-controlled phase 2 study. Lancet 2015, 385, 1198–1205. [Google Scholar] [CrossRef]
- Chiba, K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol. Therapeut. 2005, 108, 308–319. [Google Scholar] [CrossRef]
- Chubanov, V.; Mederos y Schnitzler, M.; Meißner, M.; Schäfer, S.; Abstiens, K.; Hofmann, T.; Gudermann, T. Natural and synthetic modulators of SK (K(ca)2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. Br. J. Pharmacol. 2012, 166, 1357–1376. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Yue, Z.; Sun, B.; Yang, W.; Xie, J.; Ni, E.; Feng, Y.; Mahmood, R.; Zhang, Y.; Yue, L. Sphingosine and FTY720 are potent inhibitors of the transient receptor potential melastatin 7 (TRPM7) channels. Br. J. Pharmacol. 2013, 168, 1294–1312. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Molecular Target | Manipulation | Potential Drug | Main Outcomes | Key Evidence |
|---|---|---|---|---|
| HIF-1α and HIF-2α signaling | Downregulation of HIF-1α and NADPH 2 oxidase;Upregulation of HIF-2α and Sod2 | Not identified | Hypertension and SDB | 1- HIF-2α+/− mice have an increased HIF-1α expression and consequent activation of Nox2 transcription in CB, while the reduced cellular oxidation in HIF-1α+/− mice is caused by an elevated expression of HIF-2α and Sod2 gene [203]; 2- HIF-2α+/− mice had decreased gene expression of Sod2, increased HVR, breathing instability and post-sigh apneas, and elevated blood pressure. The treatment with the antioxidant MnTMPyP abolished the autonomic and ventilatory dysfunctions [201]; 3- HIF-1α+/− mice are resistant to IH-induced LTF in CB, augmented HVR, and hypertension [202]. |
| Gasotransmitters | Blockade of CSE | 2-Arylidene Hydrazinecarbodithioates | 1- 2-Arylidene Hydrazinecarbodithioates is a potent and selective inhibitor of CSE [214]; 2- CSE−/− mice have a blunted CB sensory activity and impaired HVR [204]; 3- L-PAG, a blocker of CSE, reduces H2S levels in the CB by 55% and abolishes hypoxia-evoked H2S generation [204]; 4- L-PAG reduced the release of catecholamines from adrenal medulla [204], decreased apneas in HO-2−/− mice in a dose-dependent manner [206], and normalized blood pressure in SH rats [53]. | |
| Purinergic system | Antagonism of P2X2/3 receptors | AF-219 and AF-454 | 1- AF-219 and AF-454 are highly selective P2X3 receptor antagonists and AF-219 was clinically tested to treat patients with refractory chronic cough [215]; 2- In SH rats, AF-219 administered to the CB reduced the blood pressure in a dose-dependent manner and decreased the sympathetic tone [212]; 3- Systemic administration of AF-454 blunted the HVR and reduced the occurrence of apneas in newborn rats [213]. | |
| Leptin-TRPM7 axis | Blockade of TRPM7 channels | FTY720 | 1- FTY720 is a fingolimod that downregulates sphingosine-1 phosphate receptor and is an FDA approved drug for treating multiple sclerosis [216]; 2- FTY720 is a potent inhibitor of TRPM7 channels [217,218] and prevents the leptin-induced increase in TRPM7 currents in glomus cells [96]; 3- FTY720 administered to the CB abolished hypertension in C57BL/6J mice under leptin infusion [96]; 4- Leptin increases ventilation and HVR. FTY720 could stabilize breathing and treat SDB (?). |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, L.J.; Polotsky, V.Y. Carotid Body and Metabolic Syndrome: Mechanisms and Potential Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 5117. https://doi.org/10.3390/ijms21145117
Kim LJ, Polotsky VY. Carotid Body and Metabolic Syndrome: Mechanisms and Potential Therapeutic Targets. International Journal of Molecular Sciences. 2020; 21(14):5117. https://doi.org/10.3390/ijms21145117
Chicago/Turabian StyleKim, Lenise J., and Vsevolod Y. Polotsky. 2020. "Carotid Body and Metabolic Syndrome: Mechanisms and Potential Therapeutic Targets" International Journal of Molecular Sciences 21, no. 14: 5117. https://doi.org/10.3390/ijms21145117
APA StyleKim, L. J., & Polotsky, V. Y. (2020). Carotid Body and Metabolic Syndrome: Mechanisms and Potential Therapeutic Targets. International Journal of Molecular Sciences, 21(14), 5117. https://doi.org/10.3390/ijms21145117