Heterogeneous Polymer Dynamics Explored Using Static 1H NMR Spectra



Abstract
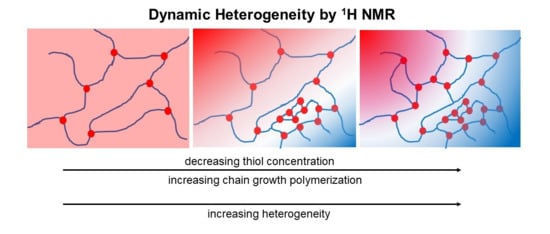
:
1. Introduction
2. Results and Discussion

2.1. Impact of Activation Energy on M2
2.2. Distributions in Dynamic Rates
2.3. Multiple Dynamic Processes
2.4. Distributions in BTT-TCDDA Networks
2.5. Dynamic Correlation Times
2.6. Arrhenius Behavior for TCDDA-BTT Networks
3. Materials and Methods
3.1. NMR Spectroscopy
3.2. Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| BTT | 1,3,5-benzenetrithiol |
| DC | Davidson-Cole |
| DMA | Dynamic mechanical analysis |
| DOAJ | Directory of open access journals |
| EEC | Entropy enthalpy compensation |
| M2 | Second moment |
| NMR | Nuclear Magnetic Resonance |
| TCDDA | Tricyclodecane dimethonal diacrylate |
| Tg | Glass transition temperature |
| Tg(NMR) | Glass transition temperature obtained from NMR M2 analysis |
| UV | Ultraviolet |
Appendix A
A.1. NMR Second Moment






A.2. Correlation Time Distributions


A.3. Internal Dynamic Impact on the Second Moment


References
- Bakhmutov, V.I. Strategies for solid-state NMR studies of materials: From diamagnetic to paramagnetic porous solids. Chem. Rev. 2011, 111, 530–562. [Google Scholar] [CrossRef] [PubMed]
- Youngman, R. NMR spectroscopy in glass science: A review of the elements. Materials 2018, 11, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, R.F.; Dawson, D.M.; Ashbrook, S.E. Exploiting NMR spectroscopy for the study of disorder in solids. Int. Rev. Phys. Chem. 2017, 36, 39–115. [Google Scholar] [CrossRef]
- Pecher, O.; Carretero-González, J.; Griffith, K.J.; Grey, C.P. Materials’ methods: NMR in battery research. Chem. Mater. 2017, 29, 213–242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Miyoshi, T.; Sun, P. (Eds.) NMR Methods for Characterization of Synthetic and Natural Polymers; Royal Society of Chemistry: London, UK, 2019; p. 565. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford University Press: New York, NY, USA, 1961; p. 599. [Google Scholar]
- Slichter, C.P. Principles of Magnetic Resonance, 3rd ed.; Springer: Berlin, Germany, 1990; p. 655. [Google Scholar]
- Gee, B.; Eckert, H. Cation distribution in mixed-alkali silicate glasses. NMR Studies by 23Na-{7Li} and 23Na-{6Li} Spin Echo double resonance. J. Phys. Chem. 1996, 100, 3705–3712. [Google Scholar] [CrossRef]
- Alam, T.M.; McLaughlin, J.; Click, C.C.; Conzone, S.; Brow, R.K.; Boyle, T.J.; Zwanziger, J.W. Investigation of sodium distribution in phosphate glasses using Spin-Echo 23Na NMR. J. Phys. Chem. B 2000, 104, 1464–1472. [Google Scholar] [CrossRef]
- De Oliveira, M.; Aitken, B.; Eckert, H. Structure of P2O5-SiO2 pure network former glasses studied by solid state NMR spectroscopy. J. Phys. Chem. C 2018, 122, 19807–19815. [Google Scholar] [CrossRef]
- Göbel, E.; Müller-Warmuth, W.; Olyschläger, H.; Dutz, H. 7Li NMR spectra, nuclear relaxation, and lithium ion motion in alkali silicate, borate, and phosphate glasses. J. Magn. Reson. 1979, 36, 371–387. [Google Scholar] [CrossRef]
- Ratai, E.; Janssen, M.; Eckert, H. Spatial distributions and chemical environments of cations in single- and mixed alkali borate glasses: Evidence from solid state NMR. Solid State Ionics 1998, 105, 25–37. [Google Scholar] [CrossRef]
- Slichter, W.P.; Mandell, E.R. Molecular motion in some glassy polymers. J. Appl. Phys. 1959, 30, 1473–1478. [Google Scholar] [CrossRef]
- McBrierty, V.J.; McDonald, I.R. NMR of oriented polymers: The effects of molecular motion on the second and fourth moments of drawn polyoxymethylene. J. Phys. D Appl. Phys. 1973, 6, 131–143. [Google Scholar] [CrossRef]
- Urin, J.; Murín, J.; Ševčovič, L.; Chodák, I. Temperature variations of NMR second moments for drawn tapes based on polypropylene and polyethylene. Macromol. Symp. 2001, 170, 123–129. [Google Scholar] [CrossRef]
- Murin, J. Second moment of NMR spectra for oriented partially crystalline polymers. Czech. J. Phys. B 1981, 31, 62–71. [Google Scholar] [CrossRef]
- Rachocki, A.; Tritt-Goc, J.; Piślewski, N. NMR study of molecular dynamics in selected hydrophilic polymers. Solid State Nucl. Magn. Reson. 2004, 25, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Nozirov, F.; Nazirov, A.; Jurga, S.; Fu, R. Molecular dynamics of poly(l-lactide) biopolymer studied by wide-line solid-state 1H and 2H NMR spectroscopy. Solid State Nucl. Magn. Reson. 2006, 29, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.M.; Jones, B.H. Investigating chain dynamics in highly crosslinked polymers using solid-state 1H NMR spectroscopy. J. Polym. Sci. Part B Polym. Phys. 2019, 57, 1143–1156. [Google Scholar] [CrossRef]
- Jones, B.H.; Alam, T.M.; Lee, S.; Celina, M.C.; Allers, J.P.; Park, S.; Chen, L.; Martinez, E.J.; Unangst, J.L. Curing behavior, chain dynamics, and microstructure of high Tg thiol-acrylate networks with systematically varied network heterogeneity. Polymer 2020. In Press. [Google Scholar] [CrossRef]
- Lu, H.; Carioscia, J.A.; Stansbury, J.W.; Bowman, C.N. Investigations of step-growth thiol-ene polymerizations for novel dental restoratives. Dental Mater. 2005, 21, 1129–1136. [Google Scholar] [CrossRef]
- Cook, W.D.; Chen, F.; Pattison, D.W.; Hopson, P.; Beaujon, M. Thermal polymerization of thiol–ene network-forming systems. Polym. Int. 2007, 56, 1572–1579. [Google Scholar] [CrossRef]
- Senyurt, A.F.; Wei, H.; Hoyle, C.E.; Piland, S.G.; Gould, T.E. Ternary thiol−ene/acrylate photopolymers: Effect of acrylate structure on mechanical properties. Macromolecules 2007, 40, 4901–4909. [Google Scholar] [CrossRef]
- McNair, O.D.; Janisse, A.P.; Krzeminski, D.E.; Brent, D.E.; Gould, T.E.; Rawlins, J.W.; Savin, D.A. Impact properties of thiol–ene networks. ACS Appl. Mater. Interf. 2013, 5, 11004–11013. [Google Scholar] [CrossRef]
- Shelkovnikov, V.V.; Ektova, L.V.; Orlova, N.A.; Ogneva, L.N.; Derevyanko, D.I.; Shundrina, I.K.; Salnikov, G.E.; Yanshole, L.V. Synthesis and thermomechanical properties of hybrid photopolymer films based on the thiol-siloxane and acrylate oligomers. J. Mater. Sci. 2015, 50, 7544–7556. [Google Scholar] [CrossRef]
- Do, D.-H.; Ecker, M.; Voit, W.E. Characterization of a thiol-ene/acrylate-based polymer for neuroprosthetic implants. ACS Omega 2017, 2, 4604–4611. [Google Scholar] [CrossRef]
- Li, C.; Johansson, M.; Sablong, R.J.; Koning, C.E. High performance thiol-ene thermosets based on fully bio-based poly(limonene carbonate)s. Eur. Polym. J. 2017, 96, 337–349. [Google Scholar] [CrossRef]
- Cordes, A.L.; Merkel, D.R.; Patel, V.J.; Courtney, C.; McBride, M.; Yakacki, C.M.; Frick, C.P. Mechanical characterization of polydopamine-assisted silver deposition on thiol-ene polymer substrates. Surf. Coat. Techn. 2019, 358, 136–143. [Google Scholar] [CrossRef]
- Dušek, K.; Prins, W. Structure and elasticity of non-crystalline polymer networks. Adv. Polym. Sci. 1969, 1, 1–102. [Google Scholar]
- Vogel, H. The temperature dependence law of the viscosity of fluids. Phys. Z. 1921, 22, 645–646. [Google Scholar]
- Fulcher, G.S. Analysis of recent measurements of the viscosity of glasses. J. Am. Ceram. Soc. 1925, 8, 339–355. [Google Scholar] [CrossRef]
- Tammann, G.; Hesse, W. Die abhängigkeit der viscosität von der temperatur bie unterkühlten flüssigkeiten. Z. Anorg. Allg. Chem. 1926, 156, 245–257. [Google Scholar] [CrossRef]
- Plazek, D.J.; Ngai, K.L. Correlation of polymer segmental chain dynamics with temperature-dependent time-scale shifts. Macromolecules 1991, 24, 1222–1224. [Google Scholar] [CrossRef]
- Roland, C.M.; Ngai, K.L. Segmental relaxation and molecular structure in polybutadienes and polyisoprene. Macromolecules 1991, 24, 5315–5319. [Google Scholar] [CrossRef] [Green Version]
- Roland, C.M.; Ngai, K.L. Normalization of the temperature dependence of segmental relaxation times. Macromolecules 1992, 25, 5765–5768. [Google Scholar] [CrossRef] [Green Version]
- Chernov, V.M.; Fedotov, V.D. Nuclear magnetic relaxation and the type of distribution of correlation times on segmental motion in rubber. Polym. Sci. USSR 1981, 23, 1042–1054. [Google Scholar] [CrossRef]
- Connor, T.M. Distributions of correlation times and their effect on the comparison of molecular motions derived from nuclear spin-lattice and dielectric relaxation. Trans. Faraday Soc. 1964, 60, 1574–1591. [Google Scholar] [CrossRef]
- Kashiwabara, H.; Shimada, S.; Hori, Y. Relaxation spectrometry in solid polymers based in NMR and EPR measurements. Makromol. Chem. Macromol. Symp. 1990, 34, 227–235. [Google Scholar] [CrossRef]
- Davidson, D.W.; Cole, R.H. Dielectric relaxation in glycerol, propylene glycol, and n-propanol. J. Chem. Phys. 1951, 19, 1484–1490. [Google Scholar] [CrossRef]
- Medycki, W.; Latanowicz, L.; Szklarz, P.; Jakubas, R. Proton dynamics at low and high temperatures in a novel ferroelectric diammonium hypodiphosphate (NH4)2H2P2O6 (ADhP) as studied by 1H spin–lattice relaxation time and second moment of NMR line. J. Magn. Reson. 2013, 231, 54–60. [Google Scholar] [CrossRef]
- Bilski, P.; Olszewski, M.; Sergeev, N.A.; Wąsicki, J. Calculation of dipolar correlation function in solids with internal mobility. Solid State Nucl. Magn. Reson. 2004, 25, 15–20. [Google Scholar] [CrossRef]
- Latanowicz, L. Spin-lattice NMR relaxation and second moment of NMR line in solids containing CH3 groups. Concepts Magn. Reson. Part A 2015, 44, 214–225. [Google Scholar] [CrossRef]
- Latanowicz, L.; Andrew, E.R.; Reynhardt, E.C. Second moment of an NMR spectrum of a solid narrowed by molecular jumps in potential wells with nonequivalent sites. J. Magn. Reson. Ser. A 1994, 107, 194–202. [Google Scholar] [CrossRef]
- Latanowicz, L.; Medycki, W.; Jakubas, R. Complex molecular dynamics of (CH3NH3)5Bi2Br11 (MAPBB) protons from NMR relaxation and second moment of NMR spectrum. J. Magn. Reson. 2011, 211, 207–216. [Google Scholar] [CrossRef]
- Latanowicz, L.; Reynhardt, E.C. Dipolar NMR spectrum of a solid narrowed by a complex molecular motion. J. Magn. Reson. Ser. A 1996, 121, 23–32. [Google Scholar] [CrossRef]
- Hołderna-Natkaniec, K.; Latanowicz, L.; Medycki, W.; Świergiel, J.; Natkaniec, I. Complex dynamics of 1.3.5-trimethylbenzene-2.4.6-D3 studied by proton spin–lattice NMR relaxation and second moment of NMR line. J. Phys. Chem. Solids 2015, 77, 109–116. [Google Scholar] [CrossRef]
- Goc, R. Calculation of the NMR second moment for materials with different types of internal rotation. Solid State Nucl. Magn. Reson. 1998, 13, 55–61. [Google Scholar] [CrossRef]
- Goc, R.; Żogał, O.J.; Vuorimäki, A.H.; Ylinen, E.E. Van Vleck second moments and hydrogen diffusion in YH2.1—Measurements and simulations. Solid State Nucl. Magn. Reson. 2004, 25, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Gutowsky, H.S.; Pake, G.E. Structural investigations by means of nuclear magnetism. II. Hindered rotation in solids. J. Chem. Phys. 1950, 18, 162–170. [Google Scholar] [CrossRef]
- Bhadauriya, S.; Wang, X.; Pitliya, P.; Zhang, J.; Raghavan, D.; Bockstaller, M.R.; Stafford, C.M.; Douglas, J.F.; Karim, A. Tuning the relaxation of nanopatterned polymer films with polymer-grafted nanoparticles: Observation of entropy-enthalpy compensation. Nano Lett. 2018, 18, 7441–7447. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Douglas, J.F.; Stafford, C.M. A Wrinkling-based method for investigating glassy polymer film relaxation as a function of film thickness and temperature. J. Chem. Phys. 2017, 147, 154902. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, Q.-X. Isokinetic relationship, isoequilibrium relationship, and enthalpy−entropy compensation. Chem. Rev. 2001, 101, 673–696. [Google Scholar] [CrossRef]
- Riggleman, R.A.; Douglas, J.F.; De Pablo, J.J. Antiplasticization and the elastic properties of glass-forming polymer liquids. Soft Matter 2010, 6, 292–304. [Google Scholar] [CrossRef]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Fragility of glass-forming polymer liquids. J. Phys. Chem. B 2005, 109, 21350–21356. [Google Scholar] [CrossRef] [PubMed]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Generalized entropy theory of polymer glass formation. In Advances in Chemical Physics; Rice, S.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 125–222. [Google Scholar]
- Moura Ramos, J.J.; Mano, J.F.; Sauer, B.B. Some comments on the significance of the compensation effect observed in thermally stimulated current experiments. Polymer 1997, 38, 1081–1089. [Google Scholar] [CrossRef]
- Lacabanne, C.; Lamure, A.; Teyssedre, G.; Bernes, A.; Mourgues, M. Study of cooperative relaxation modes in complex systems by thermally stimulated current spectroscopy. J. Non Cryst. Solids 1994, 172–174, 884–890. [Google Scholar] [CrossRef]
- Dyre, J.C. A Phenomenological model for the meyer-neldel rule. J. Phys. C Solid State Phys. 1986, 19, 5655–5664. [Google Scholar] [CrossRef]
- Van Vleck, J.H. The dipolar broadening of magnetic resonance lines in crystals. Phys. Rev. 1948, 74, 1168–1183. [Google Scholar] [CrossRef]
- Powles, J.G.; Gutowsky, H.S. Proton magnetic resonance of the CH3 group. III. Reorientation mechanism in solids. J. Chem. Phys. 1955, 23, 1692–1699. [Google Scholar] [CrossRef]
- Banks, L.; Ellis, B. Broad-line NMR studies of molecular motion in cured epoxy resins. J. Polym. Sci. Polym. Phys. 1982, 20, 1055–1067. [Google Scholar] [CrossRef]
- Beckmann, P.A. Spectral densities and nuclear spin relaxation in solids. Phys. Rep. 1988, 171, 85–128. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network | Tg (NMR) (K)a | Ea (kJ/mol) | τ0 (ns) | σ(Ea)b | εc |
|---|---|---|---|---|---|
| R = 0.47 | 412 | 29.9 ± 2 | 0.02 ± 0.03 | <1 | >0.4 |
| R = 0.32 | 440 | 24.7 ± 2 | 0.11 ± 0.06 | <1 | >0.4 |
| R = 0.19 | 482 | 19.8 ± 1 | 0.61 ± 0.1 | 2 | 0.35 |
| R = 0.09 | 523 | 16.7 ± 1 | 1.9 ± 0.2 | 3.5 | 0.30 |
| R = 0.00 | 560 | n.d. d | n.d. d | 5 | 0.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, T.M.; Allers, J.P.; Jones, B.H. Heterogeneous Polymer Dynamics Explored Using Static 1H NMR Spectra. Int. J. Mol. Sci. 2020, 21, 5176. https://doi.org/10.3390/ijms21155176
Alam TM, Allers JP, Jones BH. Heterogeneous Polymer Dynamics Explored Using Static 1H NMR Spectra. International Journal of Molecular Sciences. 2020; 21(15):5176. https://doi.org/10.3390/ijms21155176
Chicago/Turabian StyleAlam, Todd M., Joshua P. Allers, and Brad H. Jones. 2020. "Heterogeneous Polymer Dynamics Explored Using Static 1H NMR Spectra" International Journal of Molecular Sciences 21, no. 15: 5176. https://doi.org/10.3390/ijms21155176
APA StyleAlam, T. M., Allers, J. P., & Jones, B. H. (2020). Heterogeneous Polymer Dynamics Explored Using Static 1H NMR Spectra. International Journal of Molecular Sciences, 21(15), 5176. https://doi.org/10.3390/ijms21155176