Regeneration of Damaged Tendon-Bone Junctions (Entheses)—TAK1 as a Potential Node Factor

 ,
,
Abstract
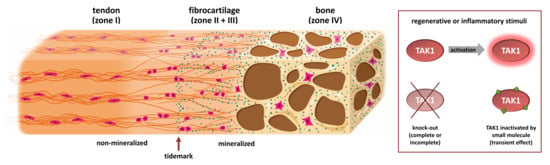
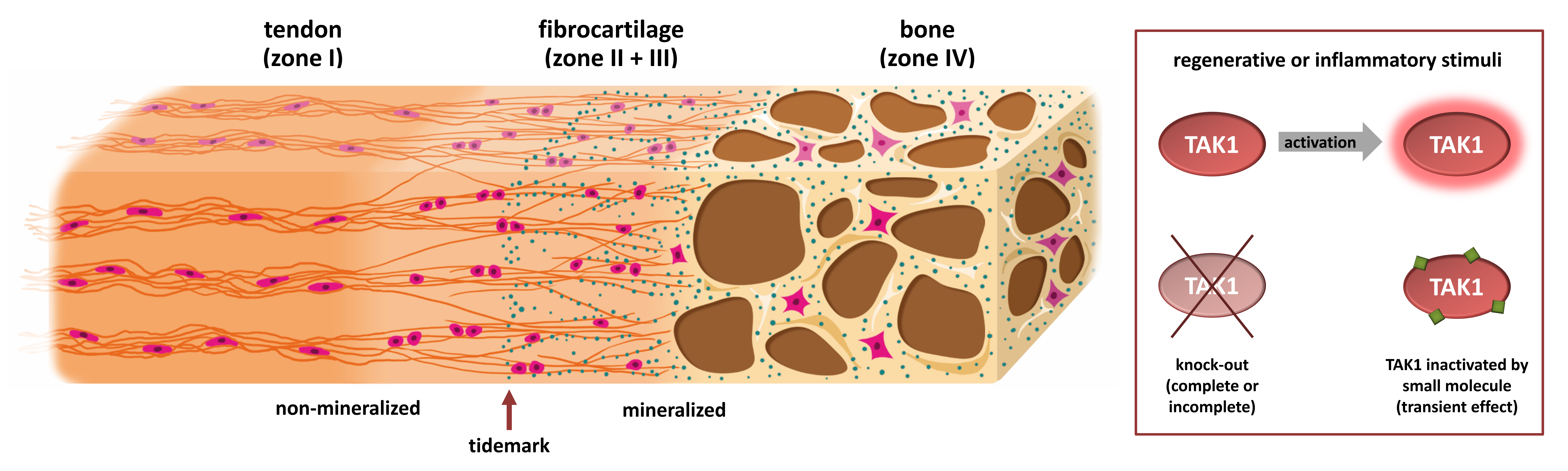
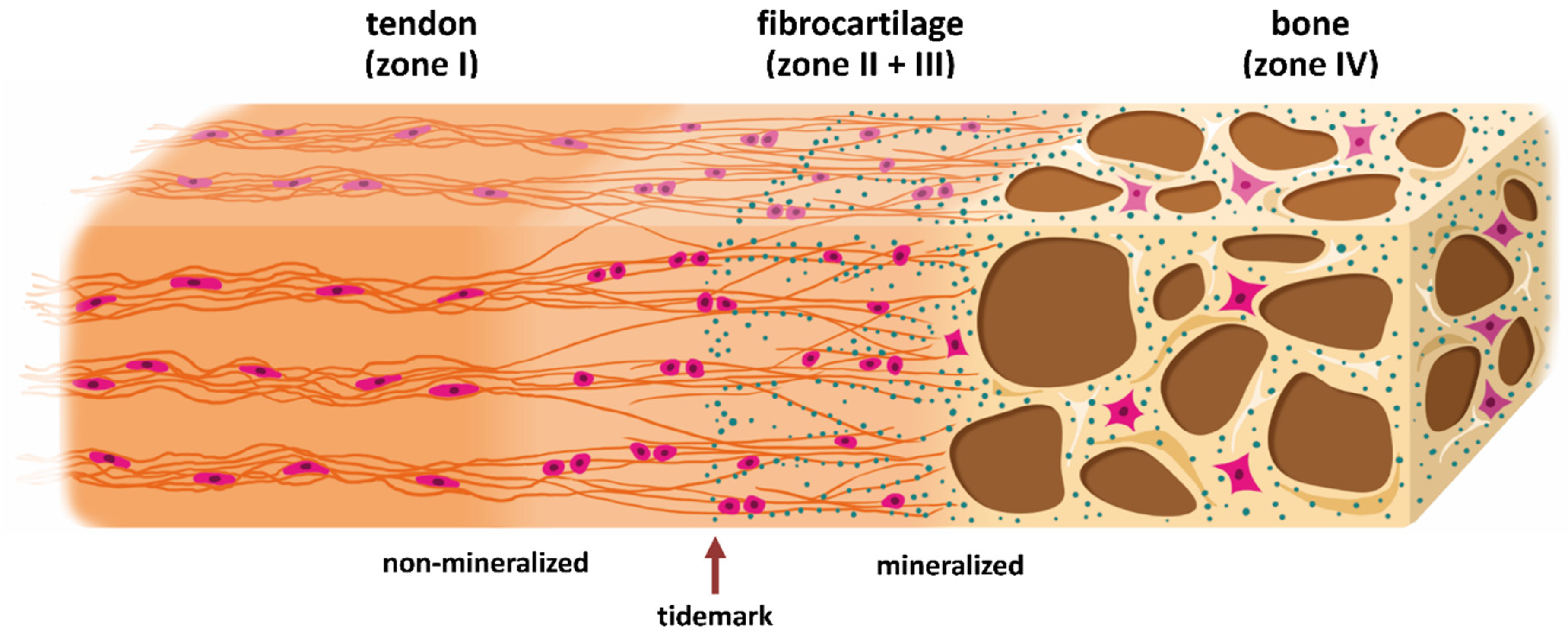
:
1. Introduction
1.1. Two Types of Entheses
1.2. Development of the Enthesis
2. Approaches towards Restoration or Regeneration of a Damaged Enthesis
3. Inflammation and the Immune System at Entheses
4. TAK1 (Transforming Growth Factor-β Activated Kinase 1)
4.1. Inhibitors of TAK1 Enzymatic Activity
4.2. TAK1 in Musculoskeletal Tissues
4.3. TAK1 in Immune Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BCR | B cell receptor |
| BMP | Bone morphogenetic protein |
| DFG | Asparagine-phenylalanine-glycine |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HH | Hedgehog |
| IC50 | Half maximal inhibitory concentration |
| IHH | Indian hedgehog |
| IL | Interleukin |
| IL1RN | Interleukin-1 antagonist (gene and cDNA) |
| IL-23R | Interleukin-23 receptor |
| JNK | c-Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| MAP3K7 | Mitogen-activated protein kinase kinase kinase 7 (also known as TAK1) |
| MAPK | Mitogen-activated protein kinase |
| M-CSF | Macrophage colony-stimulating factor |
| MMP | Matrix metalloprotease |
| MSCs | Mesenchymal stem cells |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer cells |
| NSAID | Non-steroidal anti-inflammatory drugs |
| p38 | Protein with molecular weight 38 kDa |
| PI3K | Phosphatidylinositol 3-kinase |
| PTHrP | Parathyroid hormone-related protein |
| ROR-γt | Retinoic acid-related orphan receptor gamma T |
| RUNX-2 | Runt-related transcription factor-2 |
| SCX | Scleraxis |
| SOX9 | Sex-determining region Y-box 9 |
| TAB | TAK1 binding protein |
| TAK1 | TGF-β-activated kinase-1 |
| TCR | T cell receptor |
| TGF-β | Transforming growth factor-β |
| THP-1 | Tohoku hospital pediatrics-1 |
| TLR | Toll-like-receptor |
| TNF-α | Tumor necrosis factor-alpha |
References
- Bonnevie, E.D.; Mauck, R.L. Physiology and Engineering of the Graded Interfaces of Musculoskeletal Junctions. Annu. Rev. Biomed. Eng. 2018, 20, 403–429. [Google Scholar] [CrossRef]
- Apostolakos, J.; Durant, T.J.; Dwyer, C.R.; Russell, R.P.; Weinreb, J.H.; Alaee, F.; Beitzel, K.; McCarthy, M.B.; Cote, M.P.; Mazzocca, A.D. The enthesis: A review of the tendon-to-bone insertion. Muscl. Ligam. Tend. J. 2014, 4, 333–342. [Google Scholar] [CrossRef]
- Shaw, H.M.; Benjamin, M. Structure-function relationships of entheses in relation to mechanical load and exercise. Scand. J. Med. Sci. Sports 2007, 17, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Padulo, J.; Oliva, F.; Frizziero, A.; Maffulli, N. Basic principles and recommendations in clinical and field science research: 2018 Update. Muscle Ligaments Tendons J. 2019, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.J.; Temenoff, J.S. Engineering Orthopedic Tissue Interfaces. Tissue Eng. Part B Rev. 2009, 15, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, M.; Kumai, T.; Milz, S.; Boszczyk, B.; Boszczyk, A.; Ralphs, J.R. The skeletal attachment of tendons—Tendon ‘entheses’. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2002, 133, 931–945. [Google Scholar] [CrossRef]
- Gracey, E.; Burssens, A.; Cambré, I.; Schett, G.; Lories, R.; McInnes, I.B.; Asahara, H.; Elewaut, D. Tendon and ligament mechanical loading in the pathogenesis of inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 193–207. [Google Scholar] [CrossRef]
- Wang, I.-N.E.; Mitroo, S.; Chen, F.H.; Lu, H.H.; Doty, S.B. Age-dependent changes in matrix composition and organization at the ligament-to-bone insertion. J. Orthop. Res. 2006, 24, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, J.; Sarkar, K.; Uhthoff, H.K.; Okawara, Y.; Ooshima, A. Immunohistochemical distribution of type I, II and III collagens in the rabbit supraspinatus tendon insertion. J. Anat. 1994, 185, 279–284. [Google Scholar]
- Benjamin, M.; Ralphs, J.R. Tendons and ligaments—An overview. Histol. Histopathol. 1997, 12, 1135–1144. [Google Scholar]
- Waggett, A.D.; Ralphs, J.R.; Kwan, A.P.; Woodnutt, D.; Benjamin, M. Characterization of collagens and proteoglycans at the insertion of the human Achilles tendon. Matrix Boil. 1998, 16, 457–470. [Google Scholar] [CrossRef]
- Schett, G.; Lories, R.; D’Agostino, M.A.; Elewaut, D.; Kirkham, B.; Soriano, E.R.; McGonagle, D. Enthesitis: From pathophysiology to treatment. Nat. Rev. Rheumatol. 2017, 13, 731–741. [Google Scholar] [CrossRef]
- Wopenka, B.; Kent, A.; Pasteris, J.D.; Yoon, Y.; Thomopoulos, S. The tendon-to-bone transition of the rotator cuff: A preliminary Raman spectroscopic study documenting the gradual mineralization across the insertion in rat tissue samples. Appl. Spectrosc. 2008, 62, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Genin, G.M.; Kent, A.; Birman, V.; Wopenka, B.; Pasteris, J.D.; Marquez, P.J.; Thomopoulos, S. Functional Grading of Mineral and Collagen in the Attachment of Tendon to Bone. Biophys. J. 2009, 97, 976–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetti, L.; Kuntz, L.A.; Kunold, E.; Schock, J.; Müller, K.W.; Grabmayr, H.; Stolberg-Stolberg, J.; Pfeiffer, F.; Sieber, S.A.; Burgkart, R.; et al. The microstructure and micromechanics of the tendon–bone insertion. Nat. Mater. 2017, 16, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Deymier, A.C.; An, Y.; Boyle, J.J.; Schwartz, A.G.; Birman, V.; Genin, G.M.; Thomopoulos, S.; Barber, A.H. Micro-mechanical properties of the tendon-to-bone attachment. Acta Biomater. 2017, 56, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, M.; McGonagle, D. The anatomical basis for disease localisation in seronegative spondyloarthropathy at entheses and related sites. J. Anat. 2001, 199, 503–526. [Google Scholar] [CrossRef]
- Thomopoulos, S.; Genin, G.M.; Galatz, L.M. The development and morphogenesis of the tendon-to-bone insertion—What development can teach us about healing. J. Musculoskelet. Neuronal Interact. 2010, 10, 35–45. [Google Scholar]
- Schwartz, A.G.; Pasteris, J.D.; Genin, G.M.; Daulton, T.L.; Thomopoulos, S. Mineral Distributions at the Developing Tendon Enthesis. PLoS ONE 2012, 7, e48630. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.H.; Thomopoulos, S. Functional attachment of soft tissues to bone: Development, healing, and tissue engineering. Annu. Rev. Biomed. Eng. 2013, 15, 201–226. [Google Scholar] [CrossRef] [Green Version]
- Killian, M.L.; Thomopoulos, S. Scleraxis is required for the development of a functional tendon enthesis. FASEB J. 2015, 30, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, Y.; Takimoto, A.; Akiyama, H.; Kist, R.; Scherer, G.; Nakamura, T.; Hiraki, Y.; Shukunami, C. Scx+/Sox9+ progenitors contribute to the establishment of the junction between cartilage and tendon/ligament. Development 2013, 140, 2280–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blitz, E.; Sharir, A.; Akiyama, H.; Zelzer, E. Tendon-bone attachment unit is formed modularly by a distinct pool of Scx—and Sox9 -positive progenitors. Development 2013, 140, 2680–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blitz, E.; Viukov, S.; Sharir, A.; Shwartz, Y.; Galloway, J.L.; Pryce, B.A.; Johnson, R.L.; Tabin, C.J.; Schweitzer, R.; Zelzer, E.; et al. Bone Ridge Patterning during Musculoskeletal Assembly Is Mediated through SCX Regulation of Bmp4 at the Tendon-Skeleton Junction. Dev. Cell 2009, 17, 861–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, A.G.; Long, F.; Thomopoulos, S. Enthesis fibrocartilage cells originate from a population of Hedgehog-responsive cells modulated by the loading environment. Development 2015, 142, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyment, N.A.; Breidenbach, A.P.; Schwartz, A.G.; Russell, R.P.; Aschbacher-Smith, L.; Liu, H.; Hagiwara, Y.; Jiang, R.; Thomopoulos, S.; Butler, D.L.; et al. Gdf5 progenitors give rise to fibrocartilage cells that mineralize via hedgehog signaling to form the zonal enthesis. Dev. Boil. 2015, 405, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Van Donkelaar, C.; Huiskes, R. The PTHrP–Ihh Feedback Loop in the Embryonic Growth Plate Allows PTHrP to Control Hypertrophy and Ihh to Regulate Proliferation. Biomech. Model. Mechanobiol. 2006, 6, 55–62. [Google Scholar] [CrossRef]
- Wang, M.; VanHouten, J.N.; Nasiri, A.R.; Johnson, R.L.; Broadus, A.E. PTHrP regulates the modeling of cortical bone surfaces at fibrous insertion sites during growth. J. Bone Miner. Res. 2013, 28, 598–607. [Google Scholar] [CrossRef] [Green Version]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of Rate of Cartilage Differentiation by Indian Hedgehog and PTH-Related Protein. Sciences 1996, 273, 613–622. [Google Scholar] [CrossRef]
- Calejo, I.; Costa-Almeida, R.; Reis, R.L.; Gomes, M.E. Enthesis Tissue Engineering: Biological Requirements Meet at the Interface. Tissue Eng. Part B Rev. 2019, 25, 330–356. [Google Scholar] [CrossRef]
- Thomopoulos, S.; Kim, H.-M.; Rothermich, S.Y.; Biederstadt, C.; Das, R.; Galatz, L.M. Decreased muscle loading delays maturation of the tendon enthesis during postnatal development. J. Orthop. Res. 2007, 25, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Breidenbach, A.P.; Aschbacher-Smith, L.; Lu, Y.; Dyment, N.A.; Liu, C.-F.; Liu, H.; Wylie, C.; Rao, M.; Shearn, J.T.; Rowe, D.W.; et al. Ablating hedgehog signaling in tenocytes during development impairs biomechanics and matrix organization of the adult murine patellar tendon enthesis. J. Orthop. Res. 2015, 33, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bogdanowicz, D.; Erisken, C.; Lee, N.M.; Lu, H.H. Biomimetic scaffold design for functional and integrative tendon repair. J. Shoulder Elb. Surg. 2012, 21, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.H.; Subramony, S.D.; Boushell, M.K.; Zhang, X. Tissue Engineering Strategies for the Regeneration of Orthopedic Interfaces. Ann. Biomed. Eng. 2010, 38, 2142–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Berkland, C.; Detamore, M.S. Strategies and Applications for Incorporating Physical and Chemical Signal Gradients in Tissue Engineering. Tissue Eng. Part B Rev. 2008, 14, 341–366. [Google Scholar] [CrossRef] [PubMed]
- Derwin, K.A.; Galatz, L.M.; Ratcliffe, A.; Thomopoulos, S. Enthesis Repair: Challenges and Opportunities for Effective Tendon-to-Bone Healing. J. Bone Joint. Surg. Am. 2018, 100, e109. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Caldwell, J.-M.; Doty, S.B.; Levine, W.N.; Rodeo, S.; Soslowsky, L.J.; Thomopoulos, S.; Lu, H.H. Integrating soft and hard tissues via interface tissue engineering. J. Orthop. Res. 2018, 36, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Dormer, N.H.; Berkland, C.J.; Detamore, M.S. Emerging techniques in stratified designs and continuous gradients for tissue engineering of interfaces. Ann. Biomed. Eng. 2010, 38, 2121–2141. [Google Scholar] [CrossRef]
- Li, X.; Xie, J.; Lipner, J.H.; Yuan, X.; Thomopoulos, S.; Xia, Y. Nanofiber Scaffolds with Gradations in Mineral Content for Mimicking the Tendon-to-Bone Insertion Site. Nano Lett. 2009, 9, 2763–2768. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.H.; Jiang, J. Interface Tissue Engineeringand the Formulation of Multiple-Tissue Systems. Tissue Eng. I 2006, 102, 91–111. [Google Scholar] [CrossRef]
- Atala, A.; Kasper, F.K.; Mikos, A.G. Engineering Complex Tissues. Sci. Transl. Med. 2012, 4, 160rv12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikos, A.G.; Herring, S.W.; Ochareon, P.; Elisseeff, J.; Lu, H.H.; Kandel, R.; Schoen, F.J.; Toner, M.; Mooney, D.; Atala, A.; et al. Engineering Complex Tissues. Tissue Eng. 2006, 12, 3307–3339. [Google Scholar] [CrossRef] [PubMed]
- Spalazzi, J.P.; Doty, S.B.; Moffat, K.L.; Levine, W.N.; Lu, H.H. Development of controlled matrix heterogeneity on a triphasic scaffold for orthopedic interface tissue engineering. Tissue Eng. 2006, 12, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Spalazzi, J.P.; Dagher, E.; Doty, S.B.; Guo, X.E.; Rodeo, S.A.; Lu, H.H. In vivo evaluation of a multiphased scaffold designed for orthopaedic interface tissue engineering and soft tissue-to-bone integration. J. Biomed. Mater. Res. Part A 2008, 86, 1–12. [Google Scholar] [CrossRef]
- Phillips, J.E.; Burns, K.L.; Le Doux, J.M.; Guldberg, R.E.; García, A.J. Engineering graded tissue interfaces. Proc. Natl. Acad. Sci. USA 2008, 105, 12170–12175. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Salvay, D.M.; Shea, L.D. Lentivirus delivery by adsorption to tissue engineering scaffolds. J. Biomed. Mater. Res. Part A 2009, 93, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Rowland, C.; Glass, K.A.; Ettyreddy, A.R.; Gloss, C.C.; Matthews, J.R.; Huynh, N.P.; Guilak, F. Regulation of decellularized tissue remodeling via scaffold-mediated lentiviral delivery in anatomically-shaped osteochondral constructs. Biomaterials 2018, 177, 161–175. [Google Scholar] [CrossRef]
- Singh, M.; Morris, C.P.; Ellis, R.J.; Detamore, M.S.; Berkland, C.J. Microsphere-Based Seamless Scaffolds Containing Macroscopic Gradients of Encapsulated Factors for Tissue Engineering. Tissue Eng. Part C Methods 2008, 14, 299–309. [Google Scholar] [CrossRef]
- Singh, M.; Sandhu, B.; Scurto, A.M.; Berkland, C.; Detamore, M.S. Microsphere-based scaffolds for cartilage tissue engineering: Using subcritical CO2 as a sintering agent. Acta Biomater. 2010, 6, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suciati, T.; Howard, D.; Barry, J.; Everitt, N.M.; Shakesheff, K.; Rose, F.R. Zonal release of proteins within tissue engineering scaffolds. J. Mater. Sci. Mater. Electron. 2006, 17, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wenk, E.; Zhang, X.; Meinel, L.; Vunjak-Novakovic, G.; Kaplan, D.L. Growth factor gradients via microsphere delivery in biopolymer scaffolds for osteochondral tissue engineering. J. Control. Release 2009, 134, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Jin, Q.; Giannobile, W.; Ma, P.X. Nano-fibrous scaffold for controlled delivery of recombinant human PDGF-BB. J. Control. Release 2006, 112, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calejo, I.; Costa-Almeida, R.; Reis, R.L.; Gomes, M.E. A Physiology-Inspired Multifactorial Toolbox in Soft-to-Hard Musculoskeletal Interface Tissue Engineering. Trends Biotechnol. 2020, 38, 83–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julier, Z.; Park, A.; Briquez, P.S.; Martino, M.M. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 2017, 53, 13–28. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Bridgewood, C.; Sharif, K.; Sherlock, J.; Watad, A.; McGonagle, D. Interleukin-23 pathway at the enthesis: The emerging story of enthesitis in spondyloarthropathy. Immunol. Rev. 2020, 294, 27–47. [Google Scholar] [CrossRef]
- Bridgewood, C.; Watad, A.; Russell, T.; Palmer, T.M.; Marzo-Ortega, H.; Khan, A.; Millner, P.A.; Dunsmuir, R.; Rao, A.; Loughenbury, P.; et al. Identification of myeloid cells in the human enthesis as the main source of local IL-23 production. Ann. Rheum. Dis. 2019, 78, 929–933. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent Pro- and Antiinflammatory Roles for IL-23 and IL-12 in Joint Autoimmune Inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [Green Version]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Gruber, H.E.; Marrero, E.; Cox, M.; Hanley, E. Interleukin-23 is constitutively expressed in the human annulus in vivo and in vitro, and is up-regulated in vitro by TNF-α. Biotech. Histochem. 2019, 94, 540–545. [Google Scholar] [CrossRef]
- Tu, B.; Liu, S.; Liu, G.; Yan, W.; Wang, Y.; Li, Z.; Fan, C. Macrophages derived from THP-1 promote the osteogenic differentiation of mesenchymal stem cells through the IL-23/IL-23R/β-catenin pathway. Exp. Cell Res. 2015, 339, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4−CD8− entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Layh-Schmitt, G.; Colbert, R.A. The interleukin-23/interleukin-17 axis in spondyloarthritis. Curr. Opin. Rheumatol. 2008, 20, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, A.; Yevsa, T.; Worbs, T.; Lienenklaus, S.; Sandrock, I.; Oberdörfer, L.; Korn, T.; Weiss, S.; Förster, R.; Prinz, I. Interleukin-23-Dependent γ/δ T Cells Produce Interleukin-17 and Accumulate in the Enthesis, Aortic Valve, and Ciliary Body in Mice. Arthritis Rheumatol. 2016, 68, 2476–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascão, R.; Moura, R.A.; Perpétuo, I.; Canhão, H.; Vieira-Sousa, E.; Mourão, A.F.; Rodrigues, A.M.; Polido-Pereira, J.; Queiroz, M.V.; Rosário, H.S.; et al. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R196–R208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, Y.; Nakayamada, S.; Kubo, S.; Nakano, K.; Iwata, S.; Miyagawa, I.; Ma, X.; Trimova, G.; Sakata, K.; Tanaka, Y. Th22 Cells Promote Osteoclast Differentiation via Production of IL-22 in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 2901. [Google Scholar] [CrossRef] [Green Version]
- Schön, M.P.; Erpenbeck, L. The Interleukin-23/Interleukin-17 Axis Links Adaptive and Innate Immunity in Psoriasis. Front. Immunol. 2018, 9, 1323. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 1022. [Google Scholar] [CrossRef] [Green Version]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Jin, J.; Chang, M.; Nakaya, M.; Hu, H.; Zou, Q.; Zhou, X.; Brittain, G.C.; Cheng, X.; Sun, S.-C. TPL2 mediates autoimmune inflammation through activation of the TAK1 axis of IL-17 signaling. J. Exp. Med. 2014, 211, 1689–1702. [Google Scholar] [CrossRef] [Green Version]
- Wilde, J.M.; Gumucio, J.P.; Grekin, J.A.; Sarver, D.C.; Noah, A.C.; Ruehlmann, D.G.; Davis, M.E.; Bedi, A.; Mendias, C.L. Inhibition of p38 mitogen-activated protein kinase signaling reduces fibrosis and lipid accumulation after rotator cuff repair. J. Shoulder Elb. Surg. 2016, 25, 1501–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Boil. 2009, 21, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Chen, Z.; Zhao, F.; Pan, S.; Zhang, T.; Cheng, X.; Zhang, L.; Zhang, S.; Qi, J.; Li, J.; et al. Reversal of prolonged obesity-associated cerebrovascular dysfunction by inhibiting microglial Tak1. Nat. Neurosci. 2020, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bergman, M.; Lundholm, A. Mitigation of disease- and treatment-related risks in patients with psoriatic arthritis. Arthritis Res. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giani, T.; De Masi, S.; Maccora, I.; Tirelli, F.; Simonini, G.; Falconi, M.; Cimaz, R. The Influence of Overweight and Obesity on Treatment Response in Juvenile Idiopathic Arthritis. Front. Pharmacol. 2019, 10, 637. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Shirakabe, K.; Shibuya, H.; Irie, K.; Oishi, I.; Ueno, N.; Taniguchi, T.; Nishida, E.; Matsumoto, K. Identification of a Member of the MAPKKK Family as a Potential Mediator of TGF-beta Signal Transduction. Science 1995, 270, 2008–2011. [Google Scholar] [CrossRef]
- Shibuya, H.; Yamaguchi, K.; Shirakabe, K.; Tonegawa, A.; Gotoh, Y.; Ueno, N.; Irie, K.; Nishida, E.; Matsumoto, K. TAB1: An Activator of the TAK1 MAPKKK in TGF-beta Signal Transduction. Sciences 1996, 272, 1179–1182. [Google Scholar] [CrossRef]
- Yumoto, K.; Thomas, P.S.; Lane, J.; Matsuzaki, K.; Inagaki, M.; Ninomiya-Tsuji, J.; Scott, G.J.; Ray, M.K.; Ishii, M.; Maxson, R.; et al. TGF-β-activated Kinase 1 (Tak1) Mediates Agonist-induced Smad Activation and Linker Region Phosphorylation in Embryonic Craniofacial Neural Crest-derived Cells*. J. Boil. Chem. 2013, 288, 13467–13480. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.; Yumoto, K.; Azhar, M.; Ninomiya-Tsuji, J.; Inagaki, M.; Hu, Y.; Deng, C.-X.; Kim, J.; Mishina, Y.; Kaartinen, V. Tak1, Smad4 and Trim33 redundantly mediate TGF-β3 signaling during palate development. Dev. Boil. 2014, 398, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell. Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, L.; Baas, A.; Kuipers, J.; Korswagen, H.; van de Wetering, M.; Clevers, H. Wnt activates the Tak1/Nemo-like kinase pathway. J. Biol. Chem. 2004, 279, 17232–17240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.-H.; Greenblatt, M.B.; Xie, M.; Schneider, M.D.; Zou, W.; Zhai, B.; Gygi, S.; Glimcher, L.H. TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 2009, 28, 2028–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylvain-Prévost, S.; Ear, T.; Simard, F.A.; Fortin, C.F.; Dubois, C.M.; Flamand, N.; McDonald, P.P. Activation of TAK1 by Chemotactic and Growth Factors, and Its Impact on Human Neutrophil Signaling and Functional Responses. J. Immunol. 2015, 195, 5393–5403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Nasser, H.; Chihara, T.; Suzu, S. Macropinocytosis and TAK1 mediate anti-inflammatory to pro-inflammatory macrophage differentiation by HIV-1 Nef. Cell Death Dis. 2014, 5, e1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya-Tsuji, J.; Kishimoto, K.; Hiyama, A.; Inoue, J.-I.; Cao, Z.; Matsumoto, K. The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999, 398, 252–256. [Google Scholar] [CrossRef]
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor Necrosis Factor-α-induced IKK Phosphorylation of NF-κB p65 on Serine 536 Is Mediated through the TRAF2, TRAF5, and TAK1 Signaling Pathway. J. Boil. Chem. 2003, 278, 36916–36923. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Chi, H.; Xie, M.; Schneider, M.D.; Flavell, R.A. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 2006, 7, 851–858. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.-F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef]
- Ear, T.; Fortin, C.F.; Simard, F.A.; McDonald, P.P. Constitutive Association of TGF-β–Activated Kinase 1 with the IκB Kinase Complex in the Nucleus and Cytoplasm of Human Neutrophils and Its Impact on Downstream Processes. J. Immunol. 2010, 184, 3897–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venables, J.P.; Vignal, E.; Baghdiguian, S.; Fort, P.; Tazi, J. Tissue-Specific Alternative Splicing of Tak1 Is Conserved in Deuterostomes. Mol. Boil. Evol. 2011, 29, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Shao, Q.; Fan, X.; Wu, P.; Lin, W.; Wei, H.; He, F.; Jiang, Y. Regulation of Tak1 alternative splicing by splice-switching oligonucleotides. Biochem. Biophys. Res. Commun. 2018, 497, 1018–1024. [Google Scholar] [CrossRef]
- Wu, P.; Zhou, D.; Lin, W.; Li, Y.; Wei, H.; Qian, X.; Jiang, Y.; He, F. Cell-type-resolved alternative splicing patterns in mouse liver. DNA Res. 2018, 25, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
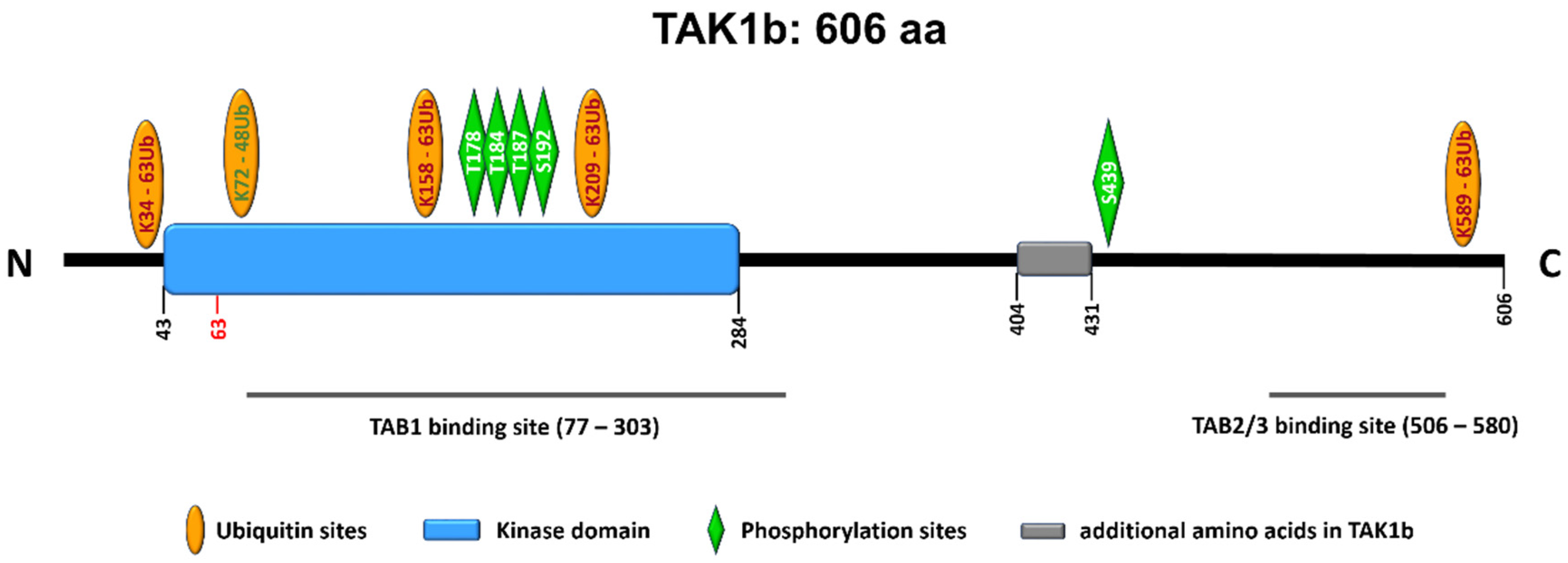
- Fan, Y.; Yu, Y.; Shi, Y.; Sun, W.; Xie, M.; Ge, N.; Mao, R.; Chang, A.; Xu, G.; Schneider, M.D.; et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J. Biol. Chem. 2010, 285, 5347–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; Von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.-H.; Landström, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nature. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef]
- Ninomiya-Tsuji, J.; Kajino, T.; Ono, K.; Ohtomo, T.; Matsumoto, M.; Shiina, M.; Mihara, M.; Tsuchiya, M.; Matsumoto, K. A Resorcylic Acid Lactone, 5Z-7-Oxozeaenol, Prevents Inflammation by Inhibiting the Catalytic Activity of TAK1 MAPK Kinase Kinase. J. Boil. Chem. 2003, 278, 18485–18490. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.-I.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, Q.; Cui, J.; Zou, J.; Xia, X.; Wang, M.; Tong, Y.; Hui, W.; Liu, D.; Su, B.; et al. TAK1 negatively regulates NF-κB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 2012, 36, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.-H.; Xiao, C.; Paschal, A.E.; Bailey, S.T.; Rao, P.; Hayden, M.S.; Lee, K.-Y.; Bussey, C.; Steckel, M.; Tanaka, N.; et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005, 19, 2668–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadrich, J.L.; O’Connor, M.B.; Coucouvanis, E. Expression of TAK1, a mediator of TGF-β and BMP signaling, during mouse embryonic development. Gene Expr. Patterns 2003, 3, 131–134. [Google Scholar] [CrossRef]
- Treiber, D.K.; Shah, N.P. Ins and outs of kinase DFG motifs. Chem. Boil. 2013, 20, 745–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Powell, F.; Larsen, N.A.; Lai, Z.; Byth, K.F.; Read, J.; Gu, R.-F.; Roth, M.; Toader, D.; Saeh, J.C.; et al. Mechanism and In Vitro Pharmacology of TAK1 Inhibition by (5Z)-7-Oxozeaenol. ACS Chem. Boil. 2013, 8, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Gurbani, D.; Weisberg, E.L.; Jones, D.S.; Rao, S.; Singer, W.D.; Bernard, F.M.; Mowafy, S.; Jenney, A.; Du, G.; et al. Studies of TAK1-centered polypharmacology with novel covalent TAK1 inhibitors. Bioorg. Med. Chem. 2016, 25, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Nomanbhoy, T.; Gurbani, D.; Patricelli, M.; Hunter, J.; Geng, J.; Herhaus, L.; Zhang, J.; Pauls, E.; Ham, Y.; et al. Discovery of Type II Inhibitors of TGFβ-Activated Kinase 1 (TAK1) and Mitogen-Activated Protein Kinase Kinase Kinase Kinase 2 (MAP4K2). J. Med. Chem. 2014, 58, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chem. Boil. 2017, 24, 1029–1039.e7. [Google Scholar] [CrossRef] [Green Version]
- Takano, S.; Uchida, K.; Itakura, M.; Iwase, D.; Aikawa, J.; Inoue, G.; Mukai, M.; Miyagi, M.; Murata, K.; Sekiguchi, H.; et al. Transforming growth factor-β stimulates nerve growth factor production in osteoarthritic synovium. BMC Musculoskelet. Disord. 2019, 20, 204. [Google Scholar] [CrossRef]
- Hsieh, H.H.S.; Agarwal, S.; Cholok, D.J.; Loder, S.J.; Kaneko, K.; Huber, A.; Chung, M.T.; Ranganathan, K.; Habbouche, J.; Li, J.; et al. Coordinating Tissue Regeneration Through Transforming Growth Factor-β Activated Kinase 1 Inactivation and Reactivation. Stem Cells 2019, 37, 766–778. [Google Scholar] [CrossRef]
- Kida, Y.; Morihara, T.; Matsuda, K.-I.; Kajikawa, Y.; Tachiiri, H.; Iwata, Y.; Sawamura, K.; Yoshida, A.; Oshima, Y.; Ikeda, T.; et al. Bone marrow-derived cells from the footprint infiltrate into the repaired rotator cuff. J. Shoulder Elb. Surg. 2013, 22, 197–205. [Google Scholar] [CrossRef]
- Yang, H.; Guo, Y.; Wang, D.; Yang, X.; Ha, C. Effect of TAK1 on osteogenic differentiation of mesenchymal stem cells by regulating BMP-2 via Wnt/β-catenin and MAPK pathway. Organogology 2018, 14, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Preobrazhenska, O.; Wodarczyk, C.; Medler, Y.; Winkel, A.; Shahab, S.; Huylebroeck, D.; Gross, G.; Verschueren, K. Transforming Growth Factor-β-activated Kinase-1 (TAK1), a MAP3K, Interacts with Smad Proteins and Interferes with Osteogenesis in Murine Mesenchymal Progenitors. J. Boil. Chem. 2005, 280, 27271–27283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, M.B.; Shim, J.-H.; Zou, W.; Sitara, D.; Schweitzer, M.; Hu, R.; Lotinun, S.; Sano, Y.; Baron, R.; Park, J.M.; et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Investig. 2010, 120, 2457–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Wang, T.; Xu, D.; Cha, B.; Liu, J.; Li, Y. Dexamethasone-induced apoptosis of osteocytic and osteoblastic cells is mediated by TAK1 activation. Biochem. Biophys. Res. Commun. 2015, 460, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Swarnkar, G.; Karuppaiah, K.; Mbalaviele, G.; Chen, T.; Abu-Amer, Y. Osteopetrosis in TAK1-deficient mice owing to defective NF-κB and NOTCH signaling. Proc. Natl. Acad. Sci. USA 2014, 112, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Gunnell, L.M.; Jonason, J.H.; Loiselle, A.E.; Kohn, A.; Schwarz, E.M.; Hilton, M.J.; O’Keefe, R.J. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J. Bone Miner. Res. 2010, 25, 1784–1797. [Google Scholar] [CrossRef] [Green Version]
- Greenblatt, M.B.; Shim, J.-H.; Glimcher, L.H. TAK1 mediates BMP signaling in cartilage. Ann. N. Y. Acad. Sci. 2010, 1192, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Hu, X.; Dai, L.; Zhang, X.; Ren, B.; Shi, W.; Liu, Z.; Duan, X.; Zhang, J.; Fu, X.; et al. Inhibition of transforming growth factor β-activated kinase 1 prevents inflammation-related cartilage degradation in osteoarthritis. Sci. Rep. 2016, 6, 34497. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K.-L. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef] [Green Version]
- Van Beuningen, H.M.; Melle, M.L.D.V.-V.; Vitters, E.L.; Schreurs, W.; Berg, W.B.V.D.; Van Osch, G.J.; Van Der Kraan, P.M. Inhibition of TAK1 and/or JAK Can Rescue Impaired Chondrogenic Differentiation of Human Mesenchymal Stem Cells in Osteoarthritis-Like Conditions. Tissue Eng. Part A 2014, 20, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Xu, M.; Bai, J.; Lin, J.; Yu, B.; Liu, Y.; Guo, X.; Shen, J.; Sun, H.; Hao, Y.; et al. Tenocyte-derived exosomes induce the tenogenic differentiation of mesenchymal stem cells through TGF-β. Cytotechnology 2019, 71, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Havis, E.; Bonnin, M.-A.; De Lima, J.E.; Charvet, B.; Milet, C.; Duprez, D. TGFβ and FGF promote tendon progenitor fate and act downstream of muscle contraction to regulate tendon differentiation during chick limb development. Developement 2016, 143, 3839–3851. [Google Scholar] [CrossRef] [Green Version]
- Barsby, T.; Guest, D.; Guest, D.J. Transforming Growth Factor Beta3 Promotes Tendon Differentiation of Equine Embryo-Derived Stem Cells. Tissue Eng. Part A 2013, 19, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Chen, X.; Li, G.; Chan, K.-M.; Heng, B.C.; Yin, Z.; Ouyang, H. Concise Review: Stem Cell Fate Guided By Bioactive Molecules for Tendon Regeneration. Stem Cells Transl. Med. 2018, 7, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.K.; Pryce, B.A.; Stabio, A.; Brigande, J.V.; Wang, C.; Xia, Z.; Tufa, S.F.; Keene, D.R.; Schweitzer, R. Tgfβ signaling is critical for maintenance of the tendon cell fate. eLife 2020, 9, 7025. [Google Scholar] [CrossRef]
- Wang, R.; Xu, B.; Xu, H.-G. Up-Regulation of TGF-β Promotes Tendon-to-Bone Healing after Anterior Cruciate Ligament Reconstruction using Bone Marrow-Derived Mesenchymal Stem Cells through the TGF-β/MAPK Signaling Pathway in a New Zealand White Rabbit Model. Cell. Physiol. Biochem. 2017, 41, 213–226. [Google Scholar] [CrossRef]
- Tsuzaki, M.; Guyton, G.; Garrett, W.; Archambault, J.M.; Herzog, W.; Almekinders, L.; Bynum, D.; Yang, X.; Banes, A.J. IL-1β induces COX2, MMP-1, -3 and -13, ADAMTS-4, IL-1β and IL-6 in human tendon cells. J. Orthop. Res. 2003, 21, 256–264. [Google Scholar] [CrossRef]
- McClellan, A.; Evans, R.; Sze, C.; Kan, S.; Paterson, Y.; Guest, D.J. A novel mechanism for the protection of embryonic stem cell derived tenocytes from inflammatory cytokine interleukin 1 beta. Sci. Rep. 2019, 9, 2755. [Google Scholar] [CrossRef]
- Richter, E.; Ventz, K.; Harms, M.; Mostertz, J.; Hochgräfe, F. Induction of Macrophage Function in Human THP-1 Cells Is Associated with Rewiring of MAPK Signaling and Activation of MAP3K7 (TAK1) Protein Kinase. Front. Cell Dev. Boil. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Chu, H.; Wang, S.; Huang, Y.; Hou, X.; Zhang, Q.; Zhou, W.; Jia, L.; Meng, Q.; Shang, L.; et al. TAK1 knock-down in macrophage alleviate lung inflammation induced by black carbon and aged black carbon. Environ. Pollut. 2019, 253, 507–515. [Google Scholar] [CrossRef]
- Chen, H.; Liu, Y.; Li, D.; Song, J.; Xia, M. PGC-1β suppresses saturated fatty acid-induced macrophage inflammation by inhibiting TAK1 activation. IUBMB Life 2016, 68, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, T.; Wieghofer, P.; Müller, P.-F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Wang, H.; Sun, X.; Liu, C.; Duan, C.; Cai, R.; Gu, X.; Zhu, S. Toll-Interleukin 1 Receptor domain-containing adaptor protein positively regulates BV2 cell M1 polarization. Eur. J. Neurosci. 2016, 43, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Courties, G.; Seiffart, V.; Presumey, J.; Escriou, V.; Scherman, D.; Zwerina, J.; Ruiz, G.; Zietara, N.; Jablonska, J.; Weiss, S.; et al. In vivo RNAi-mediated silencing of TAK1 decreases inflammatory Th1 and Th17 cells through targeting of myeloid cells. Blood 2010, 116, 3505–3516. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Chen, Y.; Lv, G.; Zhou, Z.; Mo, X.; Xie, J. Adenovirus-Mediated Small Interfering RNA Targeting TAK1 Ameliorates Joint Inflammation with Collagen-Induced Arthritis in Mice. Inflammation 2017, 40, 894–903. [Google Scholar] [CrossRef]
- Sanjo, H.; Nakayama, J.; Yoshizawa, T.; Fehling, H.J.; Akira, S.; Taki, S. Cutting Edge: TAK1 Safeguards Macrophages against Proinflammatory Cell Death. J. Immunol. 2019, 203, 783–788. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Sciences 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.; Molnár, T.; Mázló, A.; Kovács, R.; Jenei, V.; Kerekes, K.; Bácsi, A.; Koncz, G. Differences in the sensitivity of classically and alternatively activated macrophages to TAK1 inhibitor-induced necroptosis. Cancer Immunol. Immunother. 2020, 6, 212–215. [Google Scholar] [CrossRef]
- Adams, E.J.; Gu, S.; Luoma, A.M. Human gamma delta T cells: Evolution and ligand recognition. Cell. Immunol. 2015, 296, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Cuthbert, R.J.; Watad, A.; Fragkakis, E.M.; Dunsmuir, R.; Loughenbury, P.; Khan, A.; Millner, P.A.; Davison, A.; Marzo-Ortega, H.; Newton, D.; et al. Evidence that tissue resident human enthesis γδT-cells can produce IL-17A independently of IL-23R transcript expression. Ann. Rheum. Dis. 2019, 78, 1559–1565. [Google Scholar] [CrossRef]
- Hammerman, M.; Blomgran, P.; Dansac, A.; Eliasson, P.; Aspenberg, P. Different gene response to mechanical loading during early and late phases of rat Achilles tendon healing. J. Appl. Physiol. 2017, 123, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, P.; Andersson, T.; Aspenberg, P. Influence of a single loading episode on gene expression in healing rat Achilles tendons. J. Appl. Physiol. 2012, 112, 279–288. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Tsujimura, T.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Takeuchi, O.; Akira, S. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T Cells. Int. Immunol. 2006, 18, 1405–1411. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-H.; Xie, M.; Schneider, M.D.; Chen, Z.J. Essential role of TAK1 in thymocyte development and activation. Proc. Natl. Acad. Sci. USA 2006, 103, 11677–11682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, D.M.C.; Eilers, J.; Vizcaino, A.S.; Orlova, E.; Zimmermann, M.; Stanulla, M.; Schrappe, M.; Boerner, K.; Grimm, D.; Muckenthaler, M.U.; et al. MAP3K7 is recurrently deleted in pediatric T-lymphoblastic leukemia and affects cell proliferation independently of NF-κB. BMC Cancer 2018, 18, 663. [Google Scholar] [CrossRef] [PubMed]
- Schuman, J.; Chen, Y.; Podd, A.; Yu, M.; Liu, H.-H.; Wen, R.; Chen, Z.J.; Wang, D. A critical role of TAK1 in B-cell receptor–mediated nuclear factor κB activation. Blood 2009, 113, 4566–4574. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.R.; Mlodzik, M. TGF-beta activated kinase-1: New insights into the diverse roles of TAK1 in development and immunity. Cell Cycle 2006, 5, 2852–2855. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Mechanism of Inhibition | IC50 (nM) |
|---|---|---|---|
| (5Z)-7-Oxo- zeaenol (Type I) |  | Binds covalently to the cysteine 174 of the active TAK1 with DFG-in formation of the activation loop. Thus, it permanently blocks the ATP-binding pocket. | 9 |
| Takinib (Type I) |  | Slows down the auto-phosphorylation step in TAK1 activation. Competitively inhibits kinase activity via hydrogen bonds and hydrophobic interactions within the ATP-binding pocket. | 9.5 |
| NG-25 (Type II) |  | NG-25 binds to the ATP-binding pocket in the inactive DFG-out formation. It forms hydrogen bonds with different residues of the active site. | 149 |
| Compound 5 (Type I) |  | Binds covalently to the cysteine 174 of the active TAK1 with DFG-in formation of the activation loop. Thus, it permanently blocks the ATP-binding pocket. | 50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friese, N.; Gierschner, M.B.; Schadzek, P.; Roger, Y.; Hoffmann, A. Regeneration of Damaged Tendon-Bone Junctions (Entheses)—TAK1 as a Potential Node Factor. Int. J. Mol. Sci. 2020, 21, 5177. https://doi.org/10.3390/ijms21155177
Friese N, Gierschner MB, Schadzek P, Roger Y, Hoffmann A. Regeneration of Damaged Tendon-Bone Junctions (Entheses)—TAK1 as a Potential Node Factor. International Journal of Molecular Sciences. 2020; 21(15):5177. https://doi.org/10.3390/ijms21155177
Chicago/Turabian StyleFriese, Nina, Mattis Benno Gierschner, Patrik Schadzek, Yvonne Roger, and Andrea Hoffmann. 2020. "Regeneration of Damaged Tendon-Bone Junctions (Entheses)—TAK1 as a Potential Node Factor" International Journal of Molecular Sciences 21, no. 15: 5177. https://doi.org/10.3390/ijms21155177
APA StyleFriese, N., Gierschner, M. B., Schadzek, P., Roger, Y., & Hoffmann, A. (2020). Regeneration of Damaged Tendon-Bone Junctions (Entheses)—TAK1 as a Potential Node Factor. International Journal of Molecular Sciences, 21(15), 5177. https://doi.org/10.3390/ijms21155177