TRPV1 Hyperfunction Involved in Uremic Toxin Indoxyl Sulfate-Mediated Renal Tubular Damage


Abstract
:1. Introduction
2. Results
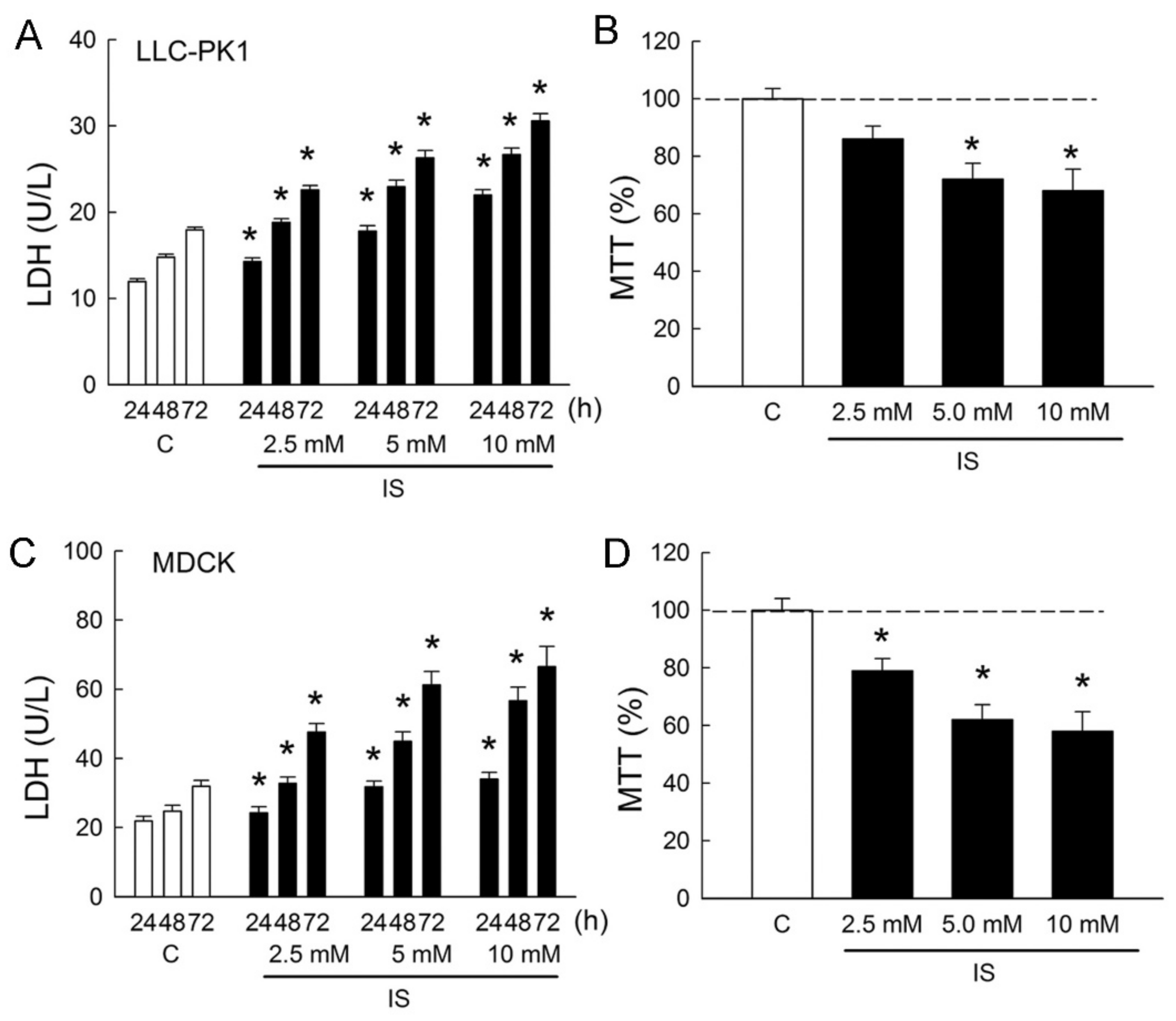
2.1. Indoxyl Sulfate Induces Renal Tubule Epithelial Cell Cytotoxicity
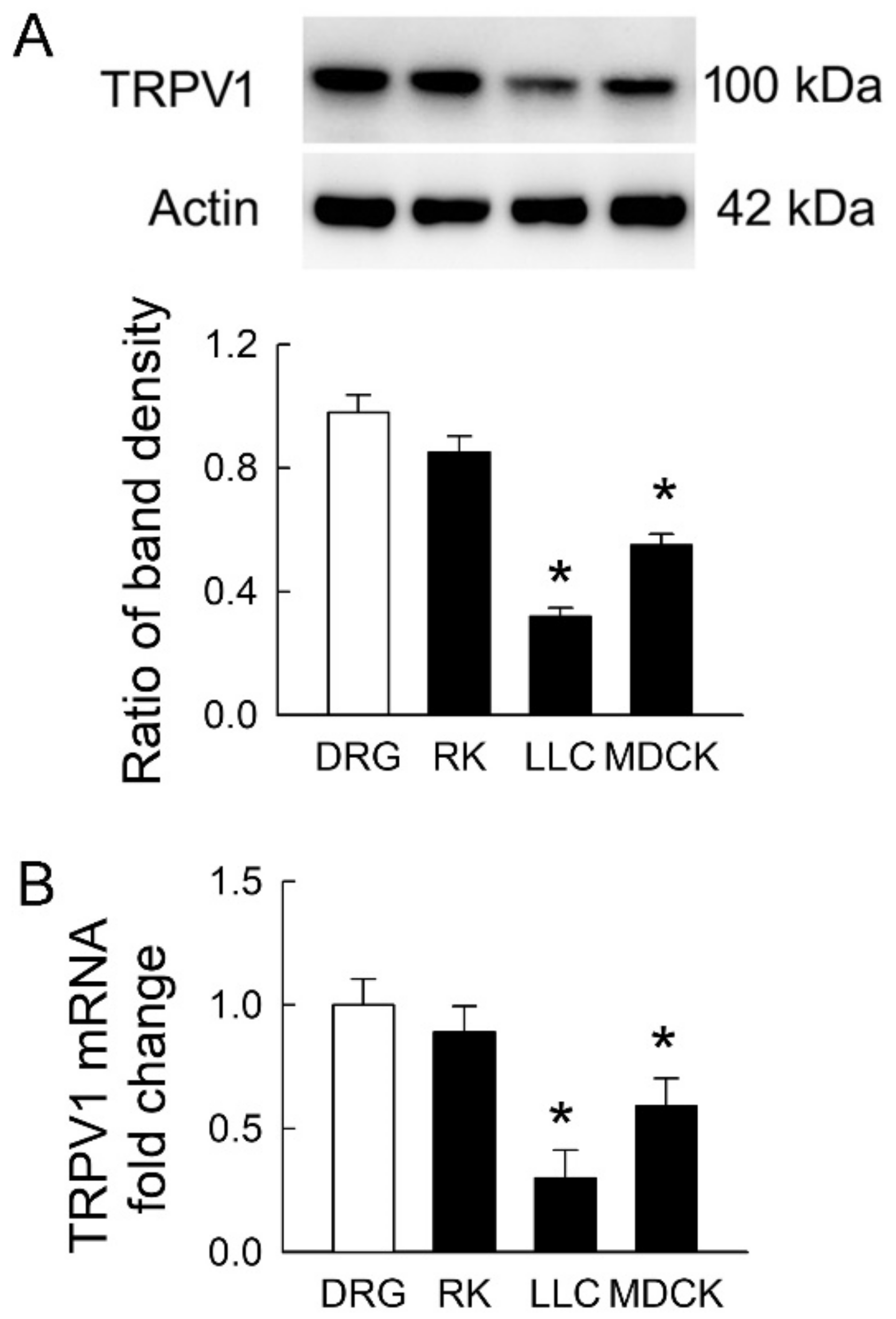
2.2. Transient Receptor Potential Vanilloid 1 (TPRV1) Is Expressed in Tubular Cell Lines
2.3. Blockade of TPRV1 Attenuates Indoxyl Sulfate (IS)-Induced Cytotoxicity
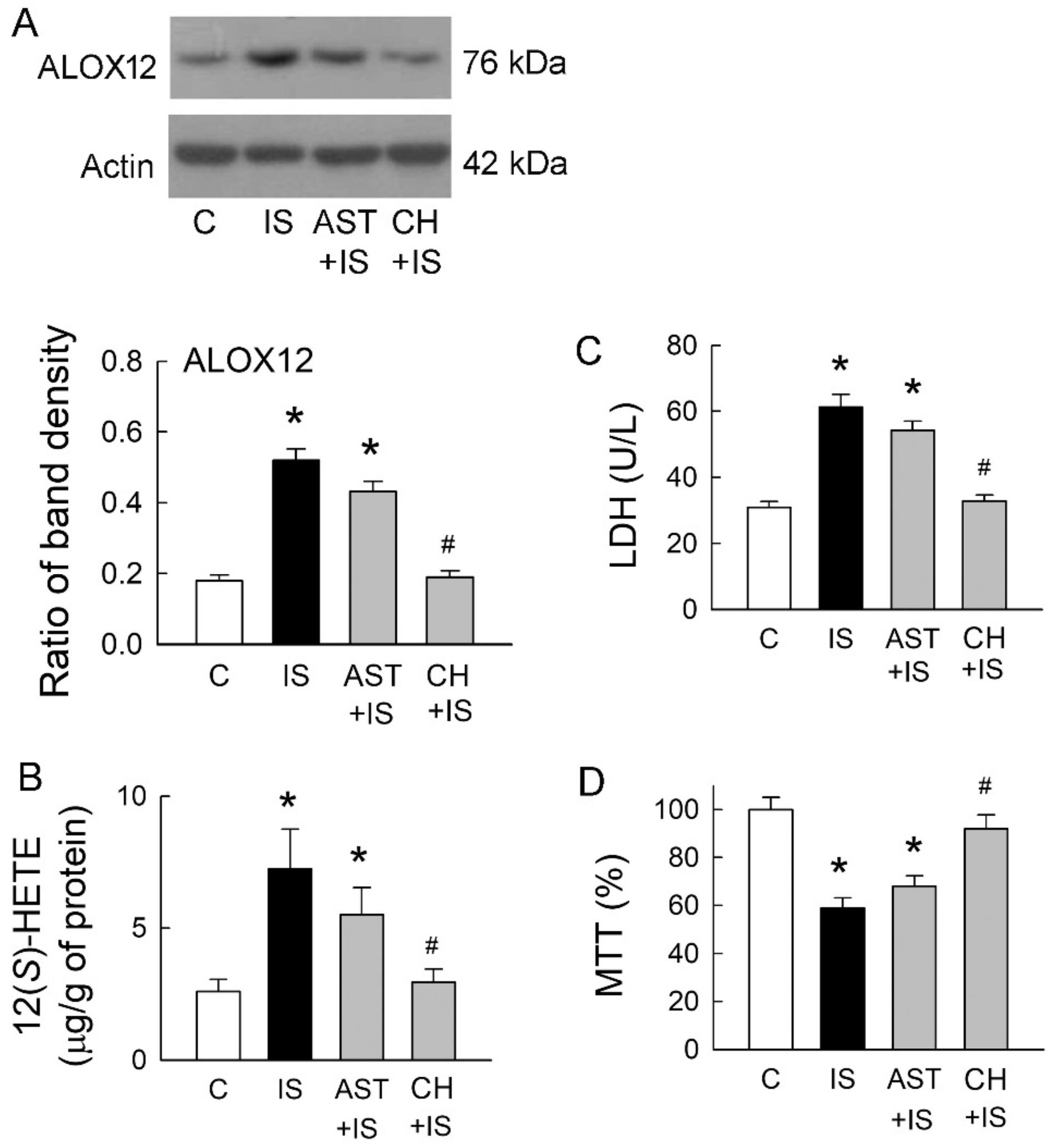
2.4. IS Enhances Arachidonate 12-Lipoxygenase (ALOX12) Expression and 12(S)-Hydroxyeicosatetraenoic Acid (HETE) Synthesis
2.5. Roles of Uremic Toxin Adsorbent and Aryl Hydrocarbon Receptor (AhR) Inhibition in Tubular Cell Injury
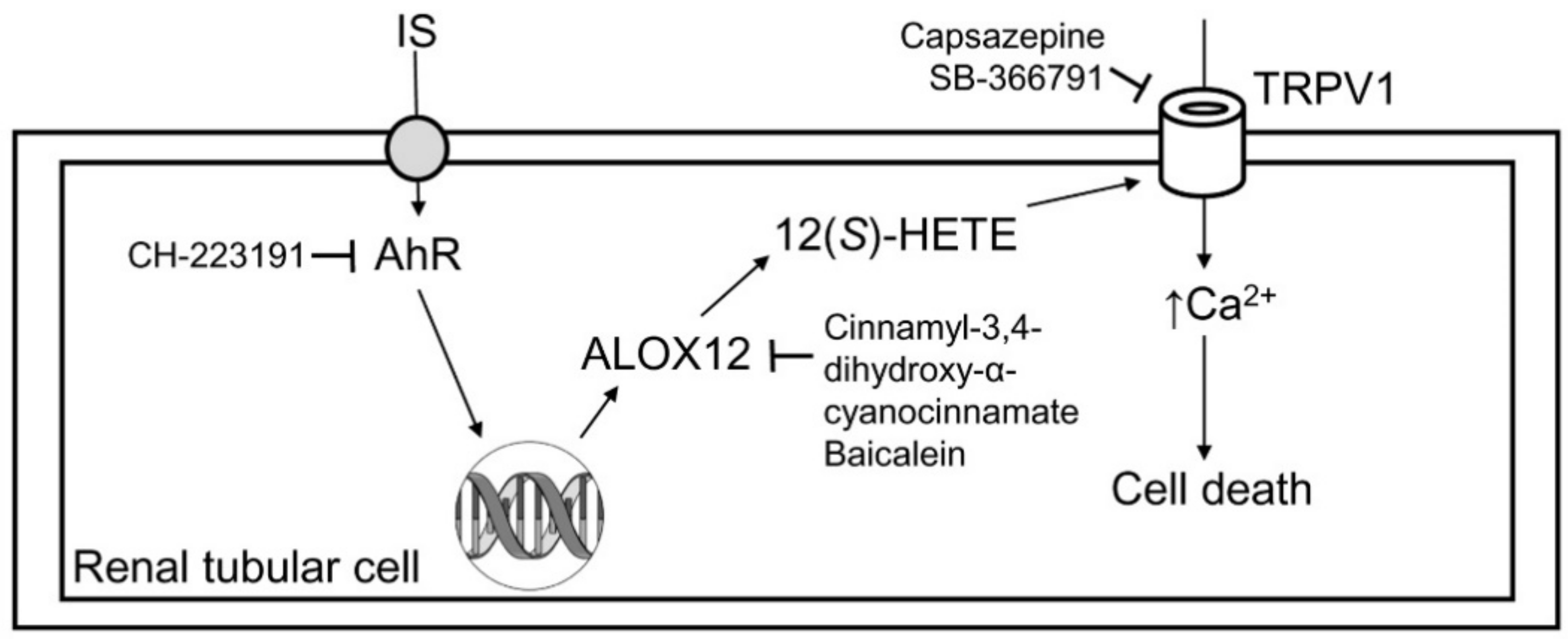
3. Discussion
4. Materials and Methods
4.1. Tubular Cell Culture and Drug Treatment
4.2. Cytotoxicity Assay
4.3. Western Blot Analysis for Protein Expression
4.4. Measurement of 12(S)-HETE Level
4.5. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) for the Quantification of TRPV1 and ALOX-12 mRNA Expression
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| ALOX12 | Arachidonate 12-lipoxygenase |
| 12(S)-HETE | 12-hydroxyeicosatetraenoic acid |
| 12(S)-HPETE | 12-hydroperoxyeicosatetraenoic acid |
| ARNA | Afferent renal nerve activity |
| AhR | Aryl hydrocarbon receptor |
| Capz | Capsazepine |
| CDC | Cinnamyl-3,4-dihydroxy-α-cyanocinnamate |
| CKD | Chronic kidney disease |
| DRG | Dorsal root ganglia |
| EMT | Epithelial-to-mesenchymal cell transition |
| IS | Indoxyl sulfate |
| LDH | Lactate dehydrogenase |
| LLC-PK1 | Porcine proximal tubular cells |
| MDCK | Madin-Darby canine kidney cells |
| OAT | Organic anion transporter |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| ROS | Reactive oxygen species |
| SB | SB-366791 |
| TRPV1 | Transient receptor potential vanilloid 1 |
References
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Liao, Q.; Chen, C.; Yang, X.; Xie, R.; Xu, J. The role of transient receptor potential vanilloid 1 in common diseases of the digestive tract and the cardiovascular and respiratory system. Front. Physiol. 2019, 10, 1064. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.; Meadows, H.J.; Gunthorpe, M.J.; Harries, M.H.; Duckworth, D.M.; Cairns, W.; Harrison, D.C.; Clarke, C.E.; Ellington, K.; Prinjha, R.K.; et al. Cloning and functional expression of a human orthologue of rat vanilloid receptor-1. Pain 2000, 88, 205–215. [Google Scholar] [CrossRef]
- Feng, N.H.; Lee, H.H.; Shiang, J.C.; Ma, M.C. Transient receptor potential vanilloid type 1 channels act as mechanoreceptors and cause substance P release and sensory activation in rat kidneys. Am. J. Physiol. Ren. Physiol. 2008, 294, F316–F325. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, D.H. Increased GFR and renal excretory function by activation of TRPV1 in the isolated perfused kidney. Pharmacol. Res. 2008, 57, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Liu, S. Role of uremic toxin indoxyl sulfate in the progression of cardiovascular disease. Life Sci. 2017, 185, 23–29. [Google Scholar] [CrossRef]
- Liu, W.C.; Tomino, Y.; Lu, K.C. Impacts of indoxyl sulfate and p-cresol sulfate on chronic kidney disease and mitigating effects of AST-120. Toxins 2018, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Choi, H.I.; Bae, E.H.; Ma, S.K.; Kim, S.W. Paricalcitol attenuates indoxyl sulfate-induced apoptosis through the inhibition of MAPK, Akt, and NF-kB activation in HK-2 cells. Korean J. Intern. Med. 2019, 34, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.J.; Small, D.M.; Ng, K.L.; Vesey, D.A.; Vitetta, L.; Francis, R.S.; Gobe, G.C.; Morais, C. Indoxyl sulfate induces apoptosis and hypertrophy in human kidney proximal tubular cells. Toxicol. Pathol. 2018, 46, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Yu, M.A.; Ryu, E.S.; Jang, Y.H.; Kang, D.H. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab. Invest. 2012, 92, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Karp, C.L. Endogenous functions of the aryl hydrocarbon receptor (AHR): Intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J. Biol. Chem. 2008, 283, 36061–36065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, W.S.; Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell-Quilley, C.P.; Lin, Y.S.; Hilchey, S.D.; Drugge, E.D.; McGiff, J.C. Renovascular actions of angiotensin II in the isolated kidney of the rat: Relationship to lipoxygenases. J. Pharmacol. Exp. Ther. 1993, 267, 676–682. [Google Scholar] [PubMed]
- Imig, J.D.; Deichmann, P.C. Afferent arteriolar responses to ANG II involve activation of PLA2 and modulation by lipoxygenase and P-450 pathways. Am. J. Physiol. 1997, 273, F274–F282. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Belusa, R.; Nowicki, S.; Aperia, A. Arachidonic acid metabolic pathways regulating activity of renal Na(+)-K(+)-ATPase are age dependent. Am. J. Physiol. Ren. Physiol. 2000, 278, F823–F829. [Google Scholar] [CrossRef]
- Lu, M.-J.; Chen, Y.-S.; Huang, H.-S.; Ma, M.-C. Hypoxic preconditioning protects rat hearts against ischemia-reperfusion injury via the arachidonate12-lipoxygenase/transient receptor potential vanilloid 1 pathway. Basic Res. Cardiol. 2014, 109, 414. [Google Scholar] [CrossRef]
- Xie, C.; Wang, D. Inhibition of renin release by arachidonic acid metabolites, 12(s)-HPETE and 12-HETE: Role of TRPV1 channels. Endocrinology 2011, 152, 3811–3819. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.Q.; Ma, S.; Wang, D.H. Activation of TRPV1 prevents salt-induced kidney damage and hypertension after renal ischemia-reperfusion injury in rats. Kidney Blood Press. Res. 2018, 43, 1285–1296. [Google Scholar] [CrossRef]
- Li, J.; Wang, D.H. Role of TRPV1 channels in renal haemodynamics and function in Dahl salt-sensitive hypertensive rats. Exp. Physiol. 2008, 93, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Tsuji, F.; Hirata, T.; Takaoka, M.; Matsumura, Y. Preventive effect of TRPV1 agonists capsaicin and resiniferatoxin on ischemia/reperfusion-induced renal injury in rats. J. Cardiovasc. Pharmacol. 2008, 51, 513–520. [Google Scholar] [CrossRef]
- Rayamajhi, S.; Contractor, T.; Wang, D.H. The potential of TRPV1 agonists for treating ischemia/reperfusion-induced renal injuries. Curr. Opin. Investig. Drugs 2009, 10, 963–970. [Google Scholar] [PubMed]
- Wang, Y.; Wang, D.H. Protective effect of TRPV1 against renal fibrosis via inhibition of TGF-β/Smad signaling in DOCA-salt hypertension. Mol. Med. 2011, 17, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taki, K.; Nakamura, S.; Miglinas, M.; Enomoto, A.; Niwa, T. Accumulation of indoxyl sulfate in OAT1/3-positive tubular cells in kidneys of patients with chronic renal failure. J. Ren. Nutr. Off. J. Counc. Ren. Nutr. Natl. Kidney Found. 2006, 16, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Ohtsuki, S.; Otagiri, M.; Takanaga, H.; Asaba, H.; Mori, S.; Terasaki, T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002, 61, 1760–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, N.; Kusuhara, H.; Sekine, T.; Endou, H.; Sugiyama, Y. Functional characterization of rat organic anion transporter 2 in LLC-PK1 cells. J. Pharmacol. Exp. Ther. 2001, 298, 1179–1184. [Google Scholar]
- Hotchkiss, A.G.; Gao, T.; Khan, U.; Berrigan, L.; Li, M.; Ingraham, L.; Pelis, R.M. Organic anion transporter 1 is inhibited by multiple mechanisms and shows a transport mode independent of exchange. Drug Metab. Dispos. 2015, 43, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, T.; Simon, S.A. Frontiers in neuroscience, TRPV1 receptors and signal transduction. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press/Taylor & Francis Group: Boca Raton, FL, USA, 2007. [Google Scholar]
- Hwang, S.; Cho, H.; Kwak, J.; Lee, S.-Y.; Kang, C.J.; Jung, S.J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhang, X.; McNaughton, P.A. Inflammatory pain: The cellular basis of heat hyperalgesia. Curr. Neuropharmacol. 2006, 4, 197–206. [Google Scholar] [CrossRef]
- Kim, S.R.; Bok, E.; Chung, Y.C.; Chung, E.S.; Jin, B.K. Interactions between CB(1) receptors and TRPV1 channels mediated by 12-HPETE are cytotoxic to mesencephalic dopaminergic neurons. Br. J. Pharmacol. 2008, 155, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecze, L.; Blum, W.; Schwaller, B. Mechanism of capsaicin receptor TRPV1-mediated toxicity in pain-sensing neurons focusing on the effects of Na+/Ca2+ fluxes and the Ca2+-binding protein calretinin. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 1680–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, C.A.; Johansen, M.E.; Lanza, D.L.; Lee, J.; Lim, J.-O.; Yost, G.S. Calcium-dependent and independent mechanisms of capsaicin receptor (TRPV1)-mediated cytokine production and cell death in human bronchial epithelial cells. J. Biochem. Mol. Toxicol. 2005, 19, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, C.A.; Taylor, J.L.; Lanza, D.L.; Carr, B.A.; Crouch, D.J.; Yost, G.S. Capsaicinoids cause inflammation and epithelial cell death through activation of vanilloid receptors. Toxicol. Sci. 2003, 73, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yang, Y.; Yang, H.; Capó-Aponte, J.E.; Tachado, S.D.; Wolosin, J.M.; Reinach, P.S. NF-κB feedback control of JNK1 activation modulates TRPV1-induced increases in IL-6 and IL-8 release by human corneal epithelial cells. Mol. Vis. 2011, 17, 3137–3146. [Google Scholar] [PubMed]
- Ma, J.; Altomare, A.; Guarino, M.; Cicala, M.; Rieder, F.; Fiocchi, C.; Li, D.; Cao, W.; Behar, J.; Biancani, P.; et al. HCl-induced and ATP-dependent upregulation of TRPV1 receptor expression and cytokine production by human esophageal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G635–G645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadoss, P.; Marcus, C.; Perdew, G.H. Role of the aryl hydrocarbon receptor in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2005, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Sheng, B.; Qiu, Y.; Yang, K.; Xiao, W.; Yang, H. Role of AhR in positive regulation of cell proliferation and survival. Cell Prolif. 2016, 49, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Niwa, T. Uremic toxicity of indoxyl sulfate. Nagoya J. Med. Sci. 2010, 72, 1–11. [Google Scholar]
- Yamaguchi, J.; Tanaka, T.; Inagi, R. Effect of AST-120 in chronic kidney disease treatment: Still a controversy? Nephron 2017, 135, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Carney, E.F. AHR activation by uraemic solutes. Nat. Rev. Nephrol. 2020, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Brito, J.S.; Borges, N.A.; Esgalhado, M.; Magliano, D.C.; Soulage, C.O.; Mafra, D. Aryl hydrocarbon receptor activation in chronic kidney disease: Role of uremic toxins. Nephron 2017, 137, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Shyu, J.F.; Lim, P.S.; Fang, T.C.; Lu, C.L.; Zheng, C.M.; Hou, Y.C.; Wu, C.C.; Lin, Y.F.; Lu, K.C. Concentration and duration of indoxyl sulfate exposure affects osteoclastogenesis by regulating NFATc1 via aryl hydrocarbon receptor. Int. J. Mol. Sci. 2020, 21, 10. [Google Scholar]
- Kim, H.Y.; Yoo, T.-H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.-Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiński, T.; Michałowska, M.; Pawlak, D. Aryl hydrocarbon receptor (AhR) and its endogenous agonist - indoxyl sulfate in chronic kidney disease. Postepy Hig. I Med. Dosw. 2017, 71, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Z.; Wang, K.; Fang, R.; Zheng, J. Expression of aryl hydrocarbon receptor in human placentas and fetal tissues. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2010, 58, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial role of the aryl hydrocarbon receptor (AhR) in indoxyl sulfate-induced vascular inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.M.; Breyer, M.D. Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int. 2007, 71, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Fu, X.; Chen, Q.; Patra, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic acid metabolism and kidney inflammation. Int. J. Mol. Sci. 2019, 20, 15. [Google Scholar] [CrossRef] [Green Version]
- Barile, F.A. Principles of Toxicology Testing; CRC Press: Boca Raton, FL, USA, 2007; p. 189. [Google Scholar]
- Adesso, S.; Paterniti, I.; Cuzzocrea, S.; Fujioka, M.; Autore, G.; Magnus, T.; Pinto, A.; Marzocco, S. AST-120 reduces neuroinflammation induced by indoxyl sulfate in glial cells. J. Clin. Med. 2018, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.C.; Wang, B.W.; Yeh, T.P.; Wu, J.L.; Chung, T.H.; Tsui, K.; Chiang, C.F.; Huang, A.J.; Huang, Y.T. Interleukin-27, a novel cytokine induced by ischemia-reperfusion injury in rat hearts, mediates cardioprotective effects via the gp130/STAT3 pathway. Basic Res. Cardiol. 2015, 110, 22. [Google Scholar] [CrossRef] [PubMed]
- Phan, W.-L.; Huang, Y.-T.; Ma, M.-C. Interleukin-27 protects cardiomyocyte-like H9c2 cells against metabolic syndrome: Role of STAT3 signaling. BioMed Res. Int. 2015, 2015, 689614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.; Wang, C.H.; Huang, S.C.; Chen, Y.W.; Yu, S.; Hwang, J.J.; Lin, J.W.; Ma, M.C.; Chen, Y.S. Novel application of amino-acid buffered solution for neuroprotection against ischemia/reperfusion injury. PLoS ONE 2019, 14, e0221039. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence |
|---|---|
| TRPV1 | 5′-GCG TTT GTC GAC TGA CTG AA-3′ (forward) 5′-CAG GAG TCC AGC TCA CCT TC-3′ (reverse) |
| ALOX12 | 5′-AGC TGG AGC CTT TCT GAC CTA TTG-3′ (forward) 5′-ACT GAT TAG GGT TGG GCA GTG TAG-3′ (reverse) |
| GAPDH | 5′-TTA GCA CCC CTG GCC AAG G-3′ (forward) 5′-CTT ACT CCT TGG AGG CCA TG-3′ (reverse) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-L.; Liao, C.-H.; Lu, K.-C.; Ma, M.-C. TRPV1 Hyperfunction Involved in Uremic Toxin Indoxyl Sulfate-Mediated Renal Tubular Damage. Int. J. Mol. Sci. 2020, 21, 6212. https://doi.org/10.3390/ijms21176212
Lu C-L, Liao C-H, Lu K-C, Ma M-C. TRPV1 Hyperfunction Involved in Uremic Toxin Indoxyl Sulfate-Mediated Renal Tubular Damage. International Journal of Molecular Sciences. 2020; 21(17):6212. https://doi.org/10.3390/ijms21176212
Chicago/Turabian StyleLu, Chien-Lin, Chun-Hou Liao, Kuo-Cheng Lu, and Ming-Chieh Ma. 2020. "TRPV1 Hyperfunction Involved in Uremic Toxin Indoxyl Sulfate-Mediated Renal Tubular Damage" International Journal of Molecular Sciences 21, no. 17: 6212. https://doi.org/10.3390/ijms21176212
APA StyleLu, C. -L., Liao, C. -H., Lu, K. -C., & Ma, M. -C. (2020). TRPV1 Hyperfunction Involved in Uremic Toxin Indoxyl Sulfate-Mediated Renal Tubular Damage. International Journal of Molecular Sciences, 21(17), 6212. https://doi.org/10.3390/ijms21176212