Investigating Developmental and Epileptic Encephalopathy Using Drosophila melanogaster


Abstract
:1. Introduction
2. Pathophysiology of Epilepsy
3. Developmental and Epileptic Encephalopathy (DEE)
3.1. Overview of DEEs
3.2. Characteristics of Monogenic DEE
4. Animal and Patient-Derived iPSC Models for Epilepsy Research
4.1. Rodent Epilepsy Models
4.2. Zebrafish Epilepsy Models
4.3. Nematode Epilepsy Models
5. Drosophila Epilepsy Models
5.1. Drosophila Models for EIEE
5.1.1. Drosophila Models Targeting Voltage-Gated Sodium Channels (Nav)
5.1.2. Drosophila Models Targeting Voltage-Gated Potassium Channel (Kv)
5.1.3. Drosophila Models Targeting Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels
5.1.4. Drosophila Models Targeting Voltage-Gated Calcium Channels (Cav)
5.1.5. Drosophila Models Targeting GABAA Receptors
5.1.6. Drosophila Models Targeting GABAB Receptors
5.1.7. Drosophila Models Targeting NMDA Receptors
5.1.8. Drosophila Models Targeting Solute Carrier Family
5.1.9. Drosophila Models Targeting Synaptic Vesicle Release/Membrane Trafficking
5.1.10. Drosophila Models Targeting Cell Adhesion Molecules
5.1.11. Drosophila Models Targeting Cytoskeletal Protein
5.1.12. Drosophila Models Targeting Intracellular Signal Transduction
5.1.13. Drosophila Models Targeting Transcription Factors
5.1.14. Drosophila Models Targeting Translation
5.1.15. Drosophila Models Targeting Post-Translational Modification
5.1.16. Drosophila Models Targeting Epigenetic Factors
5.1.17. Drosophila Models Targeting Mitochondrial Enzymes
5.1.18. Others
5.2. Drosophila Models for DEE other than EIEE
5.2.1. Drosophila Models Targeting Dynamin 1 like (DNM1L)
5.2.2. Drosophila Models Targeting Tmtc3
5.2.3. Drosophila Models Targeting Membrin
5.2.4. Drosophila Models Targeting Ube3a
5.2.5. Drosophila Models Targeting Prickle
5.3. Genetic Suppressors of Seizure Susceptibility in Drosophila
5.3.1. Genetic Screening with Bang-Sensitive Mutants
5.3.2. Genetic Screening with Drosophila Models Targeting Ube3a
5.4. Screening and Evaluation of ASDs with Drosophila Models
5.4.1. Evaluation of ASDs with Bang-Sensitive Mutants
5.4.2. Screening of ASDs Focusing on the Top1 Gene
5.4.3. Evaluation of ASDs with the PTZ-Induced Kindling Epileptogenesis Model
5.4.4. Screening of ASDs Focusing on the Pumilio Gene
6. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, N.; Nakashima, M.; Kato, M.; Heyman, E.; Inui, T.; Haginoya, K.; Watanabe, S.; Chiyonobu, T.; Morimoto, M.; Ohta, M.; et al. Detection of copy number variations in epilepsy using exome data. Clin. Genet. 2018, 93, 577–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Santiago, L.; Isom, L.L. Dravet Syndrome: A Developmental and Epileptic Encephalopathy. Epilepsy Curr. 2019, 19, 51–53. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, I.E.; Nabbout, R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 2019, 60, S17–S24. [Google Scholar] [CrossRef]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.B.S.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.A.; Wagnon, J.L.; Wengert, E.R.; Barker, B.S.; Faulkner, A.; Wagley, P.K.; Bhatia, K.; Jones, J.M.; Maniaci, M.R.; Parent, J.M.; et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 2019, 142, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Brunklaus, A.; Lal, D. Sodium channel epilepsies and neurodevelopmental disorders: From disease mechanisms to clinical application. Dev. Med. Child Neurol. 2020, 62, 784–792. [Google Scholar] [CrossRef]
- Allen, N.M.; Conroy, J.; Shahwan, A.; Lynch, B.; Correa, R.G.; Pena, S.D.J.; McCreary, D.; Magalhães, T.R.; Ennis, S.; Lynch, S.A.; et al. Unexplained early onset epileptic encephalopathy: Exome screening and phenotype expansion. Epilepsia 2016, 57, e12–e17. [Google Scholar] [CrossRef]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.F.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef] [PubMed]
- McTague, A.; Nair, U.; Malhotra, S.; Meyer, E.; Trump, N.; Gazina, E.V.; Papandreou, A.; Ngoh, A.; Ackermann, S.; Ambegaonkar, G.; et al. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology 2018, 90, e55–e66. [Google Scholar] [CrossRef] [Green Version]
- Maljevic, S.; Møller, R.S.; Reid, C.A.; Pérez-Palma, E.; Lal, D.; May, P.; Lerche, H. Spectrum of GABAA receptor variants in epilepsy. Curr. Opin. Neurol. 2019, 32, 183–190. [Google Scholar] [CrossRef]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannesen, K.M.; Nikanorova, N.; Marjanovic, D.; Pavbro, A.; Larsen, L.H.G.; Rubboli, G.; Møller, R.S. Utility of genetic testing for therapeutic decision-making in adults with epilepsy. Epilepsia 2020, 61, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; Weber, Y.G. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. 2019, 91, 90–93. [Google Scholar] [CrossRef]
- Saitsu, H.; Kato, M.; Mizuguchi, T.; Hamada, K.; Osaka, H.; Tohyama, J.; Uruno, K.; Kumada, S.; Nishiyama, K.; Nishimura, A.; et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008, 40, 782–788. [Google Scholar] [CrossRef]
- Stamberger, H.; Nikanorova, M.; Willemsen, M.H.; Accorsi, P.; Angriman, M.; Baier, H.; Benkel-Herrenbrueck, I.; Benoit, V.; Budetta, M.; Caliebe, A.; et al. STXBP1 encephalopathy A neurodevelopmental disorder including epilepsy. Neurology 2016, 86, 954–962. [Google Scholar] [CrossRef]
- Marini, C.; Mei, D.; Parmeggiani, L.; Norci, V.; Calado, E.; Ferrari, A.; Moreira, A.; Pisano, T.; Specchio, N.; Vigevano, F.; et al. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology 2010, 75, 646–653. [Google Scholar] [CrossRef]
- Homan, C.C.; Pederson, S.; To, T.H.; Tan, C.; Piltz, S.; Corbett, M.A.; Wolvetang, E.; Thomas, P.Q.; Jolly, L.A.; Gecz, J. PCDH19 regulation of neural progenitor cell differentiation suggests asynchrony of neurogenesis as a mechanism contributing to PCDH19 Girls Clustering Epilepsy. Neurobiol. Dis. 2018, 116, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ji, T.; Nelson, A.D.; Glanowska, K.; Murphy, G.G.; Jenkins, P.M.; Parent, J.M. Critical roles of αII spectrin in brain development and epileptic encephalopathy. J. Clin. Investig. 2018, 128, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Jakimiec, M.; Paprocka, J.; Śmigiel, R. CDKL5 deficiency disorder—A complex epileptic encephalopathy. Brain Sci. 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.C.; Xiong, Z.Q. Molecular and Synaptic Bases of CDKL5 Disorder. Dev. Neurobiol. 2019, 79, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Saitoh, S.; Kamei, A.; Shiraishi, H.; Ueda, Y.; Akasaka, M.; Tohyama, J.; Akasaka, N.; Hayasaka, K. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet. 2007, 81, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Olivetti, P.R.; Noebels, J.L. Interneuron, interrupted: Molecular pathogenesis of ARX mutations and X-linked infantile spasms. Curr. Opin. Neurobiol. 2012, 22, 859–865. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Dobyns, W.B. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: Proposal for a new term, “interneuronopathy”. J. Child Neurol. 2005, 20, 392–397. [Google Scholar] [CrossRef]
- Wu, T.; Yin, F.; Guang, S.; He, F.; Yang, L.; Peng, J. The Glycosylphosphatidylinositol biosynthesis pathway in human diseases. Orphanet J. Rare Dis. 2020, 15, 129. [Google Scholar] [CrossRef]
- Kato, M.; Saitsu, H.; Murakami, Y.; Kikuchi, K.; Watanabe, S.; Iai, M.; Miya, K.; Matsuura, R.; Takayama, R.; Ohba, C.; et al. PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology 2014, 82, 1587–1596. [Google Scholar] [CrossRef]
- Bayat, A.; Knaus, A.; Pendziwiat, M.; Afenjar, A.; Barakat, T.S.; Bosch, F.; Callewaert, B.; Calvas, P.; Ceulemans, B.; Chassaing, N.; et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 2020, 61, 1142–1155. [Google Scholar] [CrossRef]
- Chiyonobu, T.; Inoue, N.; Morimoto, M.; Kinoshita, T.; Murakami, Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J. Med. Genet. 2014, 51, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Osaka, H.; Murakami, Y.; Anzai, R.; Nishiyama, K.; Kodera, H.; Nakashima, M.; Tsurusaki, Y.; Miyake, N.; Kinoshita, T.; et al. PIGO mutations in intractable epilepsy and severe developmental delay with mild elevation of alkaline phosphatase levels. Epilepsia 2014, 55, e13–e17. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, D.L.; Nguyen, T.-T.-M.; Murakami, Y.; Kernohan, K.D.; Tétreault, M.; Goldsmith, C.; Doja, A.; Wagner, J.D.; Huang, L.; Hartley, T.; et al. Compound heterozygous mutations in the gene PIGP are associated with early infantile epileptic encephalopathy. Hum. Mol. Genet. 2017, 26, 1706–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunliffe, V.T.; Baines, R.A.; Giachello, C.N.G.; Lin, W.H.; Morgan, A.; Reuber, M.; Russell, C.; Walker, M.C.; Williams, R.S.B. Epilepsy research methods update: Understanding the causes of epileptic seizures and identifying new treatments using non-mammalian model organisms. Seizure 2015, 24, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grone, B.P.; Baraban, S.C. Animal models in epilepsy research: Legacies and new directions. Nat. Neurosci. 2015, 18, 339–343. [Google Scholar] [CrossRef]
- Becker, A.J. Review: Animal models of acquired epilepsy: Insights into mechanisms of human epileptogenesis. Neuropathol. Appl. Neurobiol. 2018, 44, 112–129. [Google Scholar] [CrossRef]
- Hirose, S.; Tanaka, Y.; Shibata, M.; Kimura, Y.; Ishikawa, M.; Higurashi, N.; Yamamoto, T.; Ichise, E.; Chiyonobu, T.; Ishii, A. Application of Induced Pluripotent Stem Cells in Epilepsy. Mol. Cell. Neurosci. 2020, 108, 103535. [Google Scholar] [CrossRef]
- Yoshimura, J.; Ichikawa, K.; Shoura, M.J.; Artiles, K.L.; Gabdank, I.; Wahba, L.; Smith, C.L.; Edgley, M.L.; Rougvie, A.E.; Fire, A.Z.; et al. Recompleting the Caenorhabditis elegans genome. Genome Res. 2019, 29, 1009–1022. [Google Scholar] [CrossRef] [Green Version]
- Phifer-Rixey, M.; Nachman, M.W. Insights into mammalian biology from the wild house mouse Mus musculus. Elife 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W. Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochem. Res. 2017, 42, 1873–1888. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Buckmaster, P.S.; Galanopoulou, A.S.; Moshé, S.L. Models of Seizures and Epilepsy; Elsevier Inc.: Philadelphia, PA, USA, 2005; ISBN 978-0-12-804066-9. [Google Scholar]
- Purpura, D.P.; Penry, J.; Tower, D.B.; Woodbury, D.M.; Walter, R.D. Experimental Models of Epilepsy—A Manual for the Laboratory Worker; Raven Press: New York, NY, USA, 1972; ISBN 9780911216264. [Google Scholar]
- Pinel, J.P.J.; Mucha, R.F.; Phillips, A.G. Spontaneous seizures generated in rats by kindling: A preliminary report. Physiol. Psychol. 1975, 3, 127–129. [Google Scholar] [CrossRef]
- Schauwecker, P.E. The relevance of individual genetic background and its role in animal models of epilepsy. Epilepsy Res. 2011, 97, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, W.N.; Taylor, L.; Beyer, B.; Tempel, B.L.; White, H.S. Electroconvulsive thresholds of inbred mouse strains. Genomics 2001, 74, 306–312. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Wenzel, H.J.; Robbins, C.A.; Sosunov, A.A.; Schwartzkroin, P.A. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience 2003, 122, 551–561. [Google Scholar] [CrossRef]
- Cantallops, I.; Routtenberg, A. Kainic acid induction of mossy fiber sprouting: Dependence on mouse strain. Hippocampus 2000, 10, 269–273. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Glaser, G.H. A Review of Mouse Mutants as Genetic Models of Epilepsy. Epilepsia 1985, 26, 143–150. [Google Scholar] [CrossRef]
- Fletcher, C.F.; Lutz, C.M.; O’Sullivan, T.N.; Shaughnessy, J.D.; Hawkes, R.; Frankel, W.N.; Copeland, N.G.; Jenkins, N.A. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell 1996, 87, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.L.; Baraban, S.C. Characterization of Inhibitory Circuits in the Malformed Hippocampus of Lis1 Mutant Mice. J. Neurophysiol. 2007, 98, 2737–2746. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Greenwood, J.S.F.; Calcagnotto, M.E.; Kirsch, H.E.; Barbaro, N.M.; Baraban, S.C. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann. Neurol. 2007, 61, 139–152. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.S.; McCarthy, K.D. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Nagao, Y.; Shimizu, S.; Serikawa, T.; Terada, R.; Fujimoto, M.; Okuda, A.; Mukai, T.; Sasa, M.; Kurachi, Y.; et al. Expressional analysis of inwardly rectifying Kir4.1 channels in Noda epileptic rat (NER). Brain Res. 2013, 1517, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kinboshi, M.; Shimizu, S.; Mashimo, T.; Serikawa, T.; Ito, H.; Ikeda, A.; Takahashi, R.; Ohno, Y. Down-Regulation of Astrocytic Kir4.1 Channels during the Audiogenic Epileptogenesis in Leucine-Rich Glioma-Inactivated 1 (Lgi1) Mutant Rats. Int. J. Mol. Sci. 2019, 20, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 2018, 11, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawel, K.; Langlois, M.; Martins, T.; van der Ent, W.; Tiraboschi, E.; Jacmin, M.; Crawford, A.D.; Esguerra, C.V. Seizing the moment: Zebrafish epilepsy models. Neurosci. Biobehav. Rev. 2020, 116, 1–20. [Google Scholar] [CrossRef]
- Baraban, S.C.; Taylor, M.R.; Castro, P.A.; Baier, H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience 2005, 131, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Grone, B.P.; Marchese, M.; Hamling, K.R.; Kumar, M.G.; Krasniak, C.S.; Sicca, F.; Santorelli, F.M.; Patel, M.; Baraban, S.C. Epilepsy, Behavioral Abnormalities, and Physiological Comorbidities in Syntaxin-Binding Protein 1 (STXBP1) Mutant Zebrafish. PLoS ONE 2016, 11, e0151148. [Google Scholar] [CrossRef] [Green Version]
- Fuller, T.D.; Westfall, T.A.; Das, T.; Dawson, D.V.; Slusarski, D.C. High-throughput behavioral assay to investigate seizure sensitivity in zebrafish implicates ZFHX3 in epilepsy. J. Neurogenet. 2018, 32, 92–105. [Google Scholar] [CrossRef]
- Griffin, A.; Hamling, K.R.; Knupp, K.; Hong, S.G.; Lee, L.P.; Baraban, S.C. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017, 140, 669–683. [Google Scholar] [CrossRef]
- Dinday, M.T.; Baraban, S.C. Large-scale phenotype-based antiepileptic drug screening in a zebrafish model of Dravet syndrome. eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kecskés, A.; Copmans, D.; Langlois, M.; Crawford, A.D.; Ceulemans, B.; Lagae, L.; De Witte, P.A.M.; Esguerra, C.V. Pharmacological characterization of an antisense knockdown zebrafish model of Dravet syndrome: Inhibition of epileptic seizures by the serotonin agonist fenfluramine. PLoS ONE 2015, 10, e0125898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sourbron, J.; Schneider, H.; Kecskés, A.; Liu, Y.; Buening, E.M.; Lagae, L.; Smolders, I.; De Witte, P. Serotonergic Modulation as Effective Treatment for Dravet Syndrome in a Zebrafish Mutant Model. ACS Chem. Neurosci. 2016, 7, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Griffin, A.; Hamling, K.R.; Hong, S.G.; Anvar, M.; Lee, L.P.; Baraban, S.C. Preclinical animal models for Dravet syndrome: Seizure phenotypes, comorbidities and drug screening. Front. Pharmacol. 2018, 9, 573. [Google Scholar] [CrossRef] [Green Version]
- Ceulemans, B.; Boel, M.; Leyssens, K.; Van Rossem, C.; Neels, P.; Jorens, P.G.; Lagae, L. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia 2012, 53, 1131–1139. [Google Scholar] [CrossRef]
- Tiraboschi, E.; Martina, S.; van der Ent, W.; Grzyb, K.; Gawel, K.; Cordero-Maldonado, M.L.; Poovathingal, S.K.; Heintz, S.; Satheesh, S.V.; Brattespe, J.; et al. New insights into the early mechanisms of epileptogenesis in a zebrafish model of Dravet syndrome. Epilepsia 2020, 61, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Gawel, K.; Turski, W.A.; van der Ent, W.; Mathai, B.J.; Kirstein-Smardzewska, K.J.; Simonsen, A.; Esguerra, C.V. Phenotypic Characterization of Larval Zebrafish (Danio rerio) with Partial Knockdown of the cacna1a Gene. Mol. Neurobiol. 2020, 57, 1904–1916. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Zhen, M. Action potentials drive body wall muscle contractions in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2011, 108, 2557–2562. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, J.P.; Shipley, F.B.; Linder, A.N.; Plummer, G.S.; Liu, M.; Setru, S.U.; Shaevitz, J.W.; Leifer, A.M. Whole-brain calcium imaging with cellular resolution in freely behaving Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2016, 113, E1074–E1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.; Gupta, S.; Tandon, S.; Wolkenhauer, O.; Vera, J.; Gupta, S.K. Baccoside a suppresses epileptic-like seizure/convulsion in Caenorhabditis elegans. Seizure 2010, 19, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Risley, M.G.; Kelly, S.P.; Jia, K.; Grill, B.; Dawson-Scully, K. Modulating Behavior in C. elegans Using Electroshock and Antiepileptic Drugs. PLoS ONE 2016, 11, e0163786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, M.; Yoshida, H.; Takeda, K.; Okumura, T.; Taniguchi, K.; Adachi-Yamada, T.; Tsuda, L.; Lim, Y.-M.; Dung, V.M.; Thao, D.T.P.; et al. Drosophila Models for Human Diseases; Yamaguchi, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; ISBN 978-981-13-0529-0. [Google Scholar]
- Parker, L.; Howlett, I.C.; Rusan, Z.M.; Tanouye, M.A. Seizure and Epilepsy: Studies of Seizure Disorders in. Int. Rev. Neurobiol. 2011, 99, 1–21. [Google Scholar] [CrossRef]
- Rosch, R.; Burrows, D.R.W.; Jones, L.B.; Peters, C.H.; Ruben, P.; Samarut, É. Functional Genomics of Epilepsy and Associated Neurodevelopmental Disorders Using Simple Animal Models: From Genes, Molecules to Brain Networks. Front. Cell. Neurosci. 2019, 13, 556. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, D.T.; Grigliatti, T.; Williamson, R. Temperature-sensitive mutations in Drosophila melanogaster. VII. A mutation (para-ts) causing reversible adult paralysis. Proc. Natl. Acad. Sci. USA 1971, 68, 890–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, O.; Benzer, S. Neurophysiological defects in temperature sensitive paralytic mutants of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1976, 73, 3253–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganetzky, B.; Wu, C.-F. Indirect Suppression Involving Behavioral Mutants with Altered Nerve Excitability in Drosophila melanogaster. Genetics 1982, 100, 597. [Google Scholar] [PubMed]
- Jan, Y.N.; Jan, L.Y. Genetic dissection of short-term and long-term facilitation at the Drosophila neuromuscular junction. Proc. Natl. Acad. Sci. USA 1978, 75, 515–519. [Google Scholar] [CrossRef] [Green Version]
- Parker, L.; Padilla, M.; Du, Y.; Dong, K.; Tanouye, M.A. Drosophila as a model for epilepsy: Bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics 2011, 187, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.H.; Wright, D.E.; Muraro, N.I.; Baines, R.A. Alternative splicing in the voltage-gated sodium channel DmNav regulates activation, inactivation, and persistent current. J. Neurophysiol. 2009, 102, 1994–2006. [Google Scholar] [CrossRef] [Green Version]
- Schutte, S.S.; Schutte, R.J.; Barragan, E.V.; O’Dowd, D.K. Model systems for studying cellular mechanisms of SCN1A -related epilepsy. J. Neurophysiol. 2016, 115, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loughney, K.; Kreber, R.; Ganetzky, B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell 1989, 58, 1143–1154. [Google Scholar] [CrossRef]
- Alabi, A.R.A.; Bahamonde, M.I.; Jung, H.J.; Il Kim, J.; Swartz, K.J. Portability of paddle motif function and pharmacology in voltage sensors. Nature 2007, 450, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Bosmans, F.; Martin-Eauclaire, M.F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Dib-Hajj, S.; Meisler, M.H.; Pietrobon, D. Inherited neuronal ion channelopathies: New windows on complex neurological diseases. J. Neurosci. 2008, 28, 11768–11777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, B.; Burke, B.; Pathakamuri, J.; Coleman, J.; Kuebler, D. A low-cost method for analyzing seizure-like activity and movement in Drosophila. J. Vis. Exp. 2014, 84, e51460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Khalil, B.; Ge, Q.; Desai, R.; Ryther, R.; Bazyk, A.; Bailey, R.; Haines, J.L.; Sutcliffe, J.S.; George, A.L. Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology 2001, 57, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Sugawara, T.; Mazaki-Miyazaki, E.; Takahashi, Y.; Fukushima, K.; Watanabe, M.; Hara, K.; Morikawa, T.; Yagi, K.; Yamakawa, K.; et al. Mutations of sodium channel α subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 2003, 126, 531–546. [Google Scholar] [CrossRef]
- Schutte, R.J.; Schutte, S.S.; Algara, J.; Barragan, E.V.; Gilligan, J.; Staber, C.; Savva, Y.A.; Smith, M.A.; Reenan, R.; O’Dowd, D.K. Knock-in model of Dravet syndrome reveals a constitutive and conditional reduction in sodium current. J. Neurophysiol. 2014, 112, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Gilligan, J.; Staber, C.; Schutte, R.J.; Nguyen, V.; O’Dowd, D.K.; Reenan, R. A knock-in model of human epilepsy in Drosophila reveals a novel cellular mechanism associated with heat-induced seizure. J. Neurosci. 2012, 32, 14146–14155. [Google Scholar] [CrossRef]
- Lindsay, H.A.; Baines, R.; Ffrench-Constant, R.; Lilley, K.; Jacobs, H.T.; O’Dell, K.M.C. The dominant cold-sensitive Out-cold mutants of Drosophila melanogaster have novel missense mutations in the voltage-gated sodium channel gene paralytic. Genetics 2008, 180, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aeby, A.; Sculier, C.; Bouza, A.A.; Askar, B.; Lederer, D.; Schoonjans, A.; Vander Ghinst, M.; Ceulemans, B.; Offord, J.; Lopez-Santiago, L.F.; et al. SCN1B -linked early infantile developmental and epileptic encephalopathy. Ann. Clin. Transl. Neurol. 2019, 6, 2354–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, K.; Du, Y.; Rinkevich, F.; Nomura, Y.; Xu, P.; Wang, L.; Silver, K.; Zhorov, B.S. Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem. Mol. Biol. 2014, 50, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, F.R.; Wilson, S.D.; Hall, L.M. The tip-e mutation of Drosophila decreases saxitoxin binding and interacts with other mutations affecting nerve membrane excitability. J. Neurogenet. 1986, 3, 1–17. [Google Scholar] [CrossRef]
- Ganetzky, B. Neurogenetic analysis of Drosophila mutations affecting sodium channels: Synergistic effects on viability and nerve conduction in double mutants involving tip-e. J. Neurogenet. 1986, 3, 19–31. [Google Scholar] [CrossRef]
- Papazian, D.M.; Schwarz, T.L.; Tempel, B.L.; Jan, Y.N.; Jan, L.Y. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science 1987, 237, 749–753. [Google Scholar] [CrossRef]
- Syrbe, S.; Hedrich, U.B.S.; Riesch, E.; Djémié, T.; Müller, S.; Møller, R.S.; Maher, B.; Hernandez-Hernandez, L.; Synofzik, M.; Caglayan, H.S.; et al. De novo loss-or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 2015, 47, 393–399. [Google Scholar] [CrossRef]
- Yin, X.M.; Lin, J.H.; Cao, L.; Zhang, T.M.; Zeng, S.; Zhang, K.L.; Tian, W.T.; Hu, Z.M.; Li, N.; Wang, J.L.; et al. Familial paroxysmal kinesigenic dyskinesia is associated with mutations in the KCNA1 gene. Hum. Mol. Genet. 2018, 27, 625–637. [Google Scholar] [CrossRef]
- Mosca, T.J.; Carrillo, R.A.; White, B.H.; Keshishian, H. Dissection of synaptic excitability phenotypes by using a dominant-negative Shaker K+ channel subunit. Proc. Natl. Acad. Sci. USA 2005, 102, 3477–3482. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, J.D.; Vanoye, C.G.; Kok, F.; George, A.L.; Kearney, J.A. Characterization of a KCNB1 variant associated with autism, intellectual disability, and epilepsy. Neurol. Genet. 2017, 3, e198. [Google Scholar] [CrossRef] [Green Version]
- Ueda, A.; Wu, C.F. Distinct frequency-dependent regulation of nerve terminal excitability and synaptic transmission by IA and IK potassium channels revealed by Drosophila Shaker and Shab mutations. J. Neurosci. 2006, 26, 6238–6248. [Google Scholar] [CrossRef] [PubMed]
- Ocorr, K.; Reeves, N.L.; Wessells, R.J.; Fink, M.; Chen, H.S.V.; Akasaka, T.; Yasuda, S.; Metzger, J.M.; Giles, W.; Posakony, J.W.; et al. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc. Natl. Acad. Sci. USA 2007, 104, 3943–3948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miceli, F.; Soldovieri, M.V.; Joshi, N.; Weckhuysen, S.; Cooper, E.; Taglialatela, M. KCNQ2-Related Disorders. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20437616 (accessed on 11 May 2020).
- Hegle, A.P.; Frank, C.A.; Berndt, A.; Klose, M.; Allan, D.W.; Accili, E.A. The Ih Channel Gene Promotes Synaptic Transmission and Coordinated Movement in Drosophila melanogaster. Front. Mol. Neurosci. 2017, 10, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, C.; Porro, A.; Rastetter, A.; Dalle, C.; Rivolta, I.; Bauer, D.; Oegema, R.; Nava, C.; Parrini, E.; Mei, D.; et al. HCN1 mutation spectrum: From neonatal epileptic encephalopathy to benign generalized epilepsy and beyond. Brain 2018, 141, 3160–3178. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, F.; Zou, B.; Xu, X.; Ordway, R.W. Active Zone Localization of Presynaptic Calcium Channels Encoded by the cacophony Locus of Drosophila. J. Neurosci. 2004, 24, 282–285. [Google Scholar] [CrossRef]
- Myers, C.T.; McMahon, J.M.; Schneider, A.L.; Petrovski, S.; Allen, A.S.; Carvill, G.L.; Zemel, M.; Saykally, J.E.; LaCroix, A.J.; Heinzen, E.L.; et al. De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies. Am. J. Hum. Genet. 2016, 99, 287–298. [Google Scholar] [CrossRef]
- Helbig, K.L.; Lauerer, R.J.; Bahr, J.C.; Souza, I.A.; Myers, C.T.; Uysal, B.; Schwarz, N.; Gandini, M.A.; Huang, S.; Keren, B.; et al. De Novo Pathogenic Variants in CACNA1E Cause Developmental and Epileptic Encephalopathy with Contractures, Macrocephaly, and Dyskinesias. Am. J. Hum. Genet. 2018, 103, 666–678. [Google Scholar] [CrossRef] [Green Version]
- Gorman, K.M.; Meyer, E.; Grozeva, D.; Spinelli, E.; McTague, A.; Sanchis-Juan, A.; Carss, K.J.; Bryant, E.; Reich, A.; Schneider, A.L.; et al. Bi-allelic Loss-of-Function CACNA1B Mutations in Progressive Epilepsy-Dyskinesia. Am. J. Hum. Genet. 2019, 104, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Knipple, D.C.; Soderlund, D.M. The ligand-gated chloride channel gene family of Drosophila melanogaster. Pestic. Biochem. Physiol. 2010, 97, 140–148. [Google Scholar] [CrossRef]
- Muthukumar, A.K.; Stork, T.; Freeman, M.R. Activity-dependent regulation of astrocyte GAT levels during synaptogenesis. Nat. Neurosci. 2014, 17, 1340–1350. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Miyashita, T.; Fu, T.F.; Lin, W.Y.; Wu, C.L.; Pyzocha, L.; Lin, I.R.; Saitoe, M.; Tully, T.; Chiang, A.S. NMDA receptors mediate olfactory learning and memory in Drosophila. Curr. Biol. 2005, 15, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, J.; Ueno, T.; Mitsuyoshi, M.; Kume, S.; Kume, K. The NMDA Receptor promotes sleep in the fruit fly, Drosophila melanogaster. PLoS ONE 2015, 10, e0128101. [Google Scholar] [CrossRef] [PubMed]
- Stahl, B.A.; Peco, E.; Davla, S.; Murakami, K.; Caicedo Moreno, N.A.; van Meyel, D.J.; Keene, A.C. The Taurine Transporter Eaat2 Functions in Ensheathing Glia to Modulate Sleep and Metabolic Rate. Curr. Biol. 2018, 28, 3700–3708.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hekmat-Scafe, D.S.; Lundy, M.Y.; Ranga, R.; Tanouye, M.A. Mutations in the K+/Cl- cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J. Neurosci. 2006, 26, 8943–8954. [Google Scholar] [CrossRef] [Green Version]
- Hekmat-Scafe, D.S.; Mercado, A.; Fajilan, A.A.; Lee, A.W.; Hsu, R.; Mount, D.B.; Tanouye, M.A. Seizure sensitivity is ameliorated by targeted expression of K+—Cl− cotransporter function in the mushroom body of the Drosophila brain. Genetics 2010, 184, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Heisenberg, M. Mushroom body memoir: From maps to models. Nat. Rev. Neurosci. 2003, 4, 266–275. [Google Scholar] [CrossRef]
- Rusan, Z.M.; Kingsford, O.A.; Tanouye, M.A. Modeling Glial Contributions to Seizures and Epileptogenesis: Cation-Chloride Cotransporters in Drosophila melanogaster. PLoS ONE 2014, 9, e101117. [Google Scholar] [CrossRef] [Green Version]
- Lunetti, P.; Cappello, A.R.; Marsano, R.M.; Pierri, C.L.; Carrisi, C.; Martello, E.; Caggese, C.; Dolce, V.; Capobianco, L. Mitochondrial glutamate carriers from Drosophila melanogaster: Biochemical, evolutionary and modeling studies. Biochim. Biophys. Acta 2013, 1827, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Kanellopoulos, A.K.; Mariano, V.; Spinazzi, M.; Woo, Y.J.; McLean, C.; Pech, U.; Li, K.W.; Armstrong, J.D.; Giangrande, A.; Callaerts, P.; et al. Aralar Sequesters GABA into Hyperactive Mitochondria, Causing Social Behavior Deficits. Cell 2020, 180, 1178–1197.e20. [Google Scholar] [CrossRef]
- Grigliatti, T.A.; Hall, L.; Rosenbluth, R.; Suzuki, D.T. Temperature-sensitive mutations in Drosophila melanogaster XIV. A Selection of Immobile Adults. Molec. Gen. Genet. 1973, 114, 107–114. [Google Scholar] [CrossRef]
- Van Der Bliek, A.M.; Meyerowrtz, E.M. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature 1991, 351, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Kouga, T.; Lourenço, C.M.; Shiina, M.; Goto, T.; Tsurusaki, Y.; Miyatake, S.; Miyake, N.; Saitsu, H.; Ogata, K.; et al. De novo DNM1 mutations in two cases of epileptic encephalopathy. Epilepsia 2016, 57, e18–e23. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.H.; Saito, K.; Ikeda, K. Reversible control of synaptic transmission in a single gene mutant of Drosophila melanogaster. J. Cell Biol. 1983, 96, 1517–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, J.H.; Kosaka, T.; Ikeda, K. The relationship between the number of synaptic vesicles and the amount of transmitter released. J. Neurosci. 1989, 9, 1937–1942. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, F.; Hazen, M.; Ordway, R.W. Fast synaptic fatigue in shibire mutants reveals a rapid requirement for dynamin in synaptic vesicle membrane trafficking. Nat. Neurosci. 2000, 3, 859–860. [Google Scholar] [CrossRef]
- Harrison, S.D.; Broadie, K.; van de Goor, J.; Rubin, G.M. Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron 1994, 13, 555–566. [Google Scholar] [CrossRef]
- Hosono, R.; Hekimi, S.; Kamiya, Y.; Sassa, T.; Murakami, S.; Nishiwaki, K.; Miwa, J.; Taketo, A.; Kodaira, K.-I. The unc-18 Gene Encodes a Novel Protein Affecting the Kinetics of Acetylcholine Metabolism in the Nematode Caenorhabditis elegans. J. Neurochem. 1992, 58, 1517–1525. [Google Scholar] [CrossRef]
- Wu, M.N.; Littleton, J.T.; Bhat, M.A.; Prokop, A.; Bellen, H.J. ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage-dependent manner. EMBO J. 1998, 17, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Lee, J.; Rowland, K.; Wen, Y.; Hua, H.; Carlson, N.; Lavania, S.; Parrish, J.Z.; Kim, M.D. Regulation of dendrite growth and maintenance by exocytosis. J. Cell Sci. 2015, 128, 4279–4292. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Chiyonobu, T.; Yoshida, M.; Maeda, H.; Zuiki, M.; Kidowaki, S.; Isoda, K.; Morimoto, M.; Kato, M.; Saitsu, H.; et al. Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia 2016, 57, e81–e86. [Google Scholar] [CrossRef] [Green Version]
- Huntwork, S.; Littleton, J.T. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat. Neurosci. 2007, 10, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Buhl, L.K.; Jorquera, R.A.; Akbergenova, Y.; Huntwork-Rodriguez, S.; Volfson, D.; Littleton, J.T. Differential regulation of evoked and spontaneous neurotransmitter release by C-terminal modifications of complexin. Mol. Cell. Neurosci. 2013, 52, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.W.; Buhl, L.K.; Volfson, D.; Tran, A.; Li, F.; Akbergenova, Y.; Littleton, J.T. Phosphorylation of Complexin by PKA Regulates Activity-Dependent Spontaneous Neurotransmitter Release and Structural Synaptic Plasticity. Neuron 2015, 88, 749–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowers, R.S.; Megeath, L.J.; Górska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron 2002, 36, 1063–1077. [Google Scholar] [CrossRef] [Green Version]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Mullins, C.; Hartnell, L.M.; Bonifacino, J.S. Distinct requirements for the AP-3 adaptor complex in pigment granule and synaptic vesicle biogenesis in Drosophila melanogaster. Mol. Gen. Genet. 2000, 263, 1003–1014. [Google Scholar] [CrossRef]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC 1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef]
- Fernandes, A.C.; Uytterhoeven, V.; Kuenen, S.; Wang, Y.C.; Slabbaert, J.R.; Swerts, J.; Kasprowicz, J.; Aerts, S.; Verstreken, P. Reduced synaptic vesicle protein degradation at lysosomes curbs TBC1D24/sky-induced neurodegeneration. J. Cell Biol. 2014, 207, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Fischer, B.; Lüthy, K.; Paesmans, J.; De Koninck, C.; Maes, I.; Swerts, J.; Kuenen, S.; Uytterhoeven, V.; Verstreken, P.; Versées, W. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat. Struct. Mol. Biol. 2016, 23, 965–973. [Google Scholar] [CrossRef]
- Pielage, J.; Fetter, R.D.; Davis, G.W. A postsynaptic Spectrin scaffold defines active zone size, spacing, and efficacy at the Drosophila neuromuscular junction. J. Cell Biol. 2006, 175, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, D.G.; Lee, M.K.; Feany, M.B. α-synuclein Induces Mitochondrial Dysfunction through Spectrin and the Actin Cytoskeleton. Neuron 2018, 97, 108–124.e6. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, T.; Bainton, R.J.; Fetter, R.D.; Heberlein, U.; Gaul, U. GPCR signaling is required for blood-brain barrier formation in Drosophila. Cell 2005, 123, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, L.; Sampson, C.J.; Xavier, M.J.; Bolukbasi, E.; Heck, M.M.S.; Williams, M.J. A directed miniscreen for genes involved in the Drosophila anti-parasitoid immune response. Immunogenetics 2012, 64, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.; Konrad, E.D.H.; Grüner, J.; Toutain, A.; Bok, L.A.; Cho, M.T.; Crawford, H.P.; Dubbs, H.; Douglas, G.; Jobling, R.; et al. Missense Variants in RHOBTB2 Cause a Developmental and Epileptic Encephalopathy in Humans, and Altered Levels Cause Neurological Defects in Drosophila. Am. J. Hum. Genet. 2018, 102, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Kain, P.; Chakraborty, T.S.; Sundaram, S.; Siddiqi, O.; Rodrigues, V.; Hasan, G. Reduced odor responses from antennal neurons of Gqα, phospholipase Cβ, and rdgA mutants in Drosophila support a role for a phospholipid intermediate in insect olfactory transduction. J. Neurosci. 2008, 28, 4745–4755. [Google Scholar] [CrossRef] [Green Version]
- Sampson, C.J.; Valanne, S.; Fauvarque, M.O.; Hultmark, D.; Rämet, M.; Williams, M.J. The RhoGEF Zizimin-related acts in the Drosophila cellular immune response via the Rho GTPases Rac2 and Cdc42. Dev. Comp. Immunol. 2012, 38, 160–168. [Google Scholar] [CrossRef]
- Yang, T.; Terman, J.R. 14-3-3ε Couples Protein Kinase A to Semaphorin Signaling and Silences Plexin RasGAP-Mediated Axonal Repulsion. Neuron 2012, 74, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Branco, J.; Al-Ramahi, I.; Ukani, L.; Pérez, A.M.; Fernandez-Funez, P.; Rincón-Limas, D.; Botas, J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum. Mol. Genet. 2008, 17, 376–390. [Google Scholar] [CrossRef]
- Schenck, A.; Bardoni, B.; Langmann, C.; Harden, N.; Mandel, J.-L.; Giangrande, A. CYFIP/Sra-1 Controls Neuronal Connectivity in Drosophila and Links the Rac1 GTPase Pathway to the Fragile X Protein. Neuron 2003, 38, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Brinkmann, K.; Chen, Z.; Pak, C.W.; Liao, Y.; Shi, S.; Henry, L.; Grishin, N.V.; Bogdan, S.; Rosen, M.K. The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell 2014, 156, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Abekhoukh, S.; Sahin, H.B.; Grossi, M.; Zongaro, S.; Maurin, T.; Madrigal, I.; Kazue-Sugioka, D.; Raas-Rothschild, A.; Doulazmi, M.; Carrera, P.; et al. New insights into the regulatory function of CYFIP1 in the context of WAVE- and FMRP-containing complexes. Dis. Model. Mech. 2017, 10, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Denef, N.; Schüpbach, T. The Vacuolar Proton Pump, V-ATPase, Is Required for Notch Signaling and Endosomal Trafficking in Drosophila. Dev. Cell 2009, 17, 387–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, H.K.; Jangam, S.; Tan, K.L.; Pignata, A.; Seto, E.S.; Yamamoto, S.; Wangler, M.F. A genetic screen for genes that impact peroxisomes in Drosophila identifies candidate genes for human disease. G3 Genes Genomes Genet. 2020, 10, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, T.; Tsuji, T.; Saigo, K. A concerted action of a paired-type homeobox gene, aristaless, and a homolog of Hox11/tlx homeobox gene, clawless, is essential for the distal tip development of the Drosophila leg. Dev. Biol. 2005, 279, 434–445. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, K.; Zhi, Y.; Takamura, Y.; Nagata, K.; Saigo, K.; Kojima, T.; Tanokura, M. Cooperative DNA-binding and sequence-recognition mechanism of aristaless and clawless. EMBO J. 2010, 29, 1613–1623. [Google Scholar] [CrossRef] [Green Version]
- Grueber, W.B.; Jan, L.Y.; Jan, Y.N. Different levels of the homeodomain protein cut regulate distinct dendrite branching patterns of Drosophila multidendritic neurons. Cell 2003, 112, 805–818. [Google Scholar] [CrossRef] [Green Version]
- Jinushi-Nakao, S.; Arvind, R.; Amikura, R.; Kinameri, E.; Liu, A.W.; Moore, A.W. Knot/Collier and Cut Control Different Aspects of Dendrite Cytoskeleton and Synergize to Define Final Arbor Shape. Neuron 2007, 56, 963–978. [Google Scholar] [CrossRef] [Green Version]
- Alfaiz, A.A.; Müller, V.; Boutry-Kryza, N.; Ville, D.; Guex, N.; De Bellescize, J.; Rivier, C.; Labalme, A.; Des Portes, V.; Edery, P.; et al. West syndrome caused by homozygous variant in the evolutionary conserved gene encoding the mitochondrial elongation factor GUF1. Eur. J. Hum. Genet. 2016, 24, 1001–1008. [Google Scholar] [CrossRef]
- Duan, R.; Shi, Y.; Yu, L.; Zhang, G.; Li, J.; Lin, Y.; Guo, J.; Wang, J.; Shen, L.; Jiang, H.; et al. UBA5 Mutations Cause a New Form of Autosomal Recessive Cerebellar Ataxia. PLoS ONE 2016, 11, e0149039. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Schönherr, C.; Varshney, G.K.; Dogru, M.; Hallberg, B.; Palmer, R.H. Goliath family E3 ligases regulate the recycling endosome pathway via VAMP3 ubiquitylation. EMBO J. 2013, 32, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Palmer, L.; Alexandre, C.; Kakugawa, S.; Beckett, K.; Gaugue, I.; Palmer, R.H.; Vincent, J.P. Godzilla-dependent transcytosis promotes Wingless signalling in Drosophila wing imaginal discs. Nat. Cell Biol. 2016, 18, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto-Hino, M.; Katsumata, E.; Suzuki, E.; Maeda, Y.; Kinoshita, T.; Goto, S. Nuclear envelope localization of PIG-B is essential for GPI-anchor synthesis in Drosophila. J. Cell Sci. 2018, 131, jcs218024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tea, J.S.; Luo, L. The chromatin remodeling factor Bap55 functions through the TIP60 complex to regulate olfactory projection neuron dendrite targeting. Neural Dev. 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Featherstone, D.E.; Rushton, E.; Broadie, K. Developmental regulation of glutamate receptor field size by nonvesicular glutamate release. Nat. Neurosci. 2002, 5, 141–146. [Google Scholar] [CrossRef]
- Falk, D.R.; Nash, D. Pyrimidine auxotrophy in Drosophila. Mol. Gen. Genet. MGG 1974, 131, 339–349. [Google Scholar] [CrossRef]
- Appocher, C.; Mohagheghi, F.; Cappelli, S.; Stuani, C.; Romano, M.; Feiguin, F.; Buratti, E. Major hnRNP proteins act as general TDP-43 functional modifiers both in Drosophila and human neuronal cells. Nucleic Acids Res. 2017, 45, 8026–8045. [Google Scholar] [CrossRef] [Green Version]
- O’Keefe, L.V.; Colella, A.; Dayan, S.; Chen, Q.; Choo, A.; Jacob, R.; Price, G.; Venter, D.; Richards, R.I. Drosophila orthologue of WWOX, the chromosomal fragile site FRA16D tumour suppressor gene, functions in aerobic metabolism and regulates reactive oxygen species. Hum. Mol. Genet. 2011, 20, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Morales, C.; Li, Z. Drosophila canopy b is a cochaperone of glycoprotein 93. J. Biol. Chem. 2017, 292, 6657–6666. [Google Scholar] [CrossRef] [Green Version]
- Jumbo-Lucioni, P.P.; Parkinson, W.M.; Kopke, D.L.; Broadie, K. Coordinated movement, neuromuscular synaptogenesis and trans-synaptic signaling defects in Drosophila galactosemia models. Hum. Mol. Genet. 2016, 25, 3699–3714. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.W.; Zhou, D.; Nizet, V.; Haddad, G.G. Experimental Selection for Drosophila Survival in Extremely High O2 Environments. PLoS ONE 2010, 5, e11701. [Google Scholar] [CrossRef]
- Toyoda, H.; Kinoshita-Toyoda, A.; Fox, B.; Selleck, S.B. Structural analysis of glycosaminoglycans in animals bearing mutations in sugarless, sulfateless, and tout-velu: Drosophila homologues of vertebrate genes encoding glycosaminoglycan biosynthetic enzymes. J. Biol. Chem. 2000, 275, 21856–21861. [Google Scholar] [CrossRef] [Green Version]
- Jumbo-Lucioni, P.; Parkinson, W.; Broadie, K. Overelaborated synaptic architecture and reduced synaptomatrix glycosylation in a Drosophila classic galactosemia disease model. Dis. Model. Mech. 2014, 7, 1365–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, K.R.; Yoon, Y.; Krueger, E.W.; McNiven, M.A. The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell 1999, 10, 4403–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; Van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.; Thiemann, M.; Grabenbauer, M.; Yoon, Y.; McNiven, M.A.; Schrader, M. Dynamin-like protein 1 is involved in peroxisomal fission. J. Biol. Chem. 2003, 278, 8597–8605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batzir, N.A.; Bhagwat, P.K.; Eble, T.N.; Liu, P.; Eng, C.M.; Elsea, S.H.; Robak, L.A.; Scaglia, F.; Goldman, A.M.; Dhar, S.U.; et al. De novo missense variant in the GTPase effector domain (GED) of DNM1L leads to static encephalopathy and seizures. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003673. [Google Scholar] [CrossRef] [Green Version]
- Verstreken, P.; Ly, C.V.; Venken, K.J.T.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Farhan, S.M.K.; Nixon, K.C.J.; Everest, M.; Edwards, T.N.; Long, S.; Segal, D.; Knip, M.J.; Arts, H.H.; Chakrabarti, R.; Wang, J.; et al. Identification of a novel synaptic protein, TMTC3, involved in periventricular nodular heterotopia with intellectual disability and epilepsy. Hum. Mol. Genet. 2017, 26, 4278–4289. [Google Scholar] [CrossRef]
- Praschberger, R.; Lowe, S.A.; Malintan, N.T.; Giachello, C.N.G.; Patel, N.; Houlden, H.; Kullmann, D.M.; Baines, R.A.; Usowicz, M.M.; Krishnakumar, S.S.; et al. Mutations in Membrin/GOSR2 Reveal Stringent Secretory Pathway Demands of Dendritic Growth and Synaptic Integrity. Cell Rep. 2017, 21, 97–109. [Google Scholar] [CrossRef]
- Lambrechts, R.A.; Polet, S.S.; Hernandez-Pichardo, A.; van Ninhuys, L.; Gorter, J.A.; Grzeschik, N.A.; de Koning-Tijssen, M.A.J.; de Koning, T.J.; Sibon, O.C.M. North Sea Progressive Myoclonus Epilepsy is Exacerbated by Heat, A Phenotype Primarily Associated with Affected Glia. Neuroscience 2019, 423, 1–11. [Google Scholar] [CrossRef]
- Wu, Y.; Bolduc, F.V.; Bell, K.; Tully, T.; Fang, Y.; Sehgal, A.; Fischer, J.A. A Drosophila model for Angelman syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 12399–12404. [Google Scholar] [CrossRef] [Green Version]
- Hope, K.A.; LeDoux, M.S.; Reiter, L.T. Glial overexpression of Dube3a causes seizures and synaptic impairments in Drosophila concomitant with down regulation of the Na+/K+ pump ATPα. Neurobiol. Dis. 2017, 108, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Manak, J.R.; Sowers, L.; Mei, X.; Kiyonari, H.; Abe, T.; Dahdaleh, N.S.; Yang, T.; Wu, S.; Chen, S.; et al. Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am. J. Hum. Genet. 2011, 88, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Ehaideb, S.N.; Iyengar, A.; Ueda, A.; Iacobucci Cathryn Cranston, G.J.; Bassuk, A.G.; Gubb, D.; Axelrod, J.D.; Gunawardena, S.; Wu, C.F.; Robert Manak, J. Prickle modulates microtubule polarity and axonal transport to ameliorate seizures in flies. Proc. Natl. Acad. Sci. USA 2014, 111, 11187–11192. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Parker, L.; Hormozi, L.; Tanouye, M.A. DNA topoisomerase I inhibitors ameliorate seizure-like behaviors and paralysis in a Drosophila model of epilepsy. Neuroscience 2008, 156, 722–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Tanouye, M.A. From bench to drug: Human seizure modeling using Drosophila. Prog. Neurobiol. 2008, 84, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Saras, A.; Tanouye, M.A. Mutations of the Calcium Channel Gene cacophony Suppress Seizures in Drosophila. PLoS Genet. 2016, 12, e1005784. [Google Scholar] [CrossRef] [PubMed]
- Howlett, I.C.; Rusan, Z.M.; Parker, L.; Tanouye, M.A. Drosophila as a Model for Intractable Epilepsy: Gilgamesh Suppresses Seizures in para bss1 Heterozygote Flies. G3 Genes Genomes Genet. 2013, 3, 1399–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroll, J.R.; Wong, K.G.; Siddiqui, F.M.; Tanouye, M.A. Disruption of endocytosis with the dynamin mutant shibirets1 suppresses seizures in Drosophila. Genetics 2015, 201, 1087–1102. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.A.; McGinn, A.; Reiter, L.T. A genome-wide enhancer/suppressor screen for Dube3a interacting genes in Drosophila melanogaster. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef]
- Straub, J.; Gregor, A.; Sauerer, T.; Fliedner, A.; Distel, L.; Suchy, C.; Ekici, A.B.; Ferrazzi, F.; Zweier, C. Genetic interaction screen for severe neurodevelopmental disorders reveals a functional link between Ube3a and Mef2 in Drosophila melanogaster. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, L.; Lim, Y.M. Alzheimer’s disease model system using Drosophila. In Drosophila Models for Human Diseases. Advances in Experimental Medicine and Biology; Yamaguchi, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 1076, pp. 25–40. [Google Scholar]
- Reynolds, E.R.; Stauffer, E.A.; Feeney, L.; Rojahn, E.; Jacobs, B.; McKeever, C. Treatment with the antiepileptic drugs phenytoin and gabapentin ameliorates seizure and paralysis of Drosophila bang-sensitive mutants. J. Neurobiol. 2004, 58, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hu, J.; Tanouye, M. Seizure suppression by top1 mutations in Drosophila. J. Neurosci. 2007, 27, 2927–2937. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Mohammad, F.; Sharma, A. Transcriptomic Analysis in a Drosophila Model Identifies Previously Implicated and Novel Pathways in the Therapeutic Mechanism in Neuropsychiatric Disorders. Front. Neurosci. 2011, 5, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.H.; Giachello, C.N.G.; Baines, R.A. Seizure control through genetic and pharmacological manipulation of Pumilio in Drosophila: A key component of neuronal homeostasis. Dis. Model. Mech. 2017, 10, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gennarino, V.A.; Palmer, E.E.; McDonell, L.M.; Wang, L.; Adamski, C.J.; Koire, A.; See, L.; Chen, C.A.; Schaaf, C.P.; Rosenfeld, J.A.; et al. A Mild PUM1 Mutation Is Associated with Adult-Onset Ataxia, whereas Haploinsufficiency Causes Developmental Delay and Seizures. Cell 2018, 172, 924–936.e11. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.H.; He, M.; Fan, Y.N.; Baines, R.A. An RNAi-mediated screen identifies novel targets for next-generation antiepileptic drugs based on increased expression of the homeostatic regulator pumilio. J. Neurogenet. 2018, 32, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Happ, H.C.; Carvill, G.L. A 2020 View on the Genetics of Developmental and Epileptic Encephalopathies. Epilepsy Curr. 2020, 20, 90–96. [Google Scholar] [CrossRef]
- Takata, A.; Nakashima, M.; Saitsu, H.; Mizuguchi, T.; Mitsuhashi, S.; Takahashi, Y.; Okamoto, N.; Osaka, H.; Nakamura, K.; Tohyama, J.; et al. Comprehensive analysis of coding variants highlights genetic complexity in developmental and epileptic encephalopathy. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]



{kind=link}
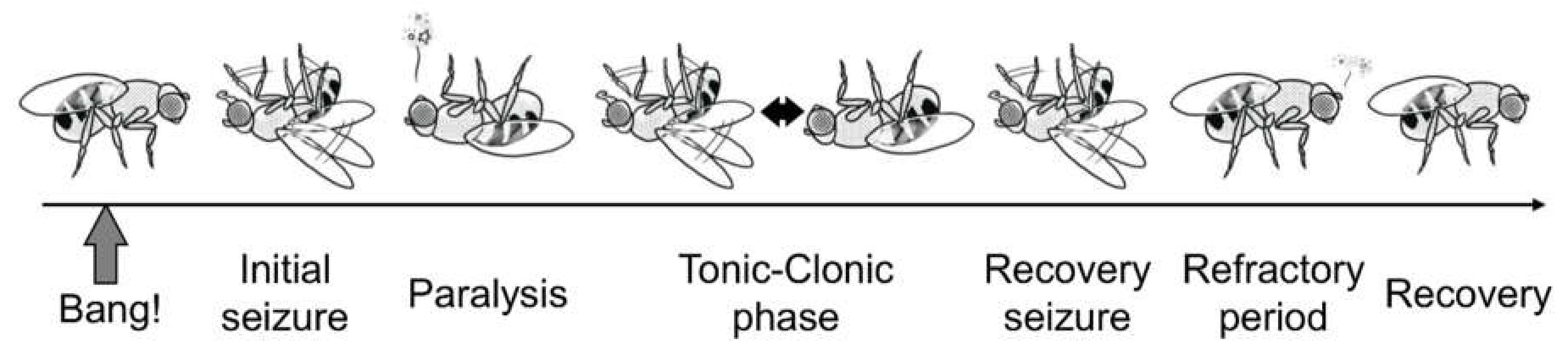{kind=link}
| Encoded Gene Function | Gene Name (Human) | EIEE# | Drosophila Orthologs | Score | ||
|---|---|---|---|---|---|---|
| Ion channel | Sodium channel | Nav1.1 | SCN1A | 6 | para | 11 of 15 |
| Nav1.2 | SCN2A | 11 | 12 of 15 | |||
| Nav1.3 | SCN3A | 62 | 11 of 15 | |||
| Nav1.6 | SCN8A | 18 | 13 of 15 | |||
| SCN1B | 52 | - (TipE/TEH1-4 *) | - | |||
| Potassium channel | Kv1.2 | KCNA2 | 32 | Sh | 12 of 15 | |
| Kv2.1 | KCNB1 | 26 | Shab | 9 of 15 | ||
| Kv7.2 | KCNQ2 | 7 | KCNQ | 10 of 15 | ||
| KCa4.1 | KCNT1 | 14 | SLO2 | 13 of 15 | ||
| KCa4.2 | KCNT2 | 57 | 12 of 15 | |||
| HCN channel | HCN1 | HCN1 | 24 | Ih | 6 of 15 | |
| Calcium channel | Cav2.1 | CACNA1A | 42 | cac | 10 of 15 | |
| Cav2.3 | CACNA1E | 69 | 10 of 15 | |||
| Neurotransmitter receptor | GABAA receptor | GABRA1 | 19 | CG8916 | 10 of 15 | |
| GABRA2 | 78 | 11 of 15 | ||||
| GABRA5 | 79 | 9 of 15 | ||||
| GABRG2 | 74 | 8 of 15 | ||||
| GABRB1 | 45 | Lcch3 | 14 of 15 | |||
| GABRB3 | 43 | 12 of 15 | ||||
| GABAB receptor | GABBR2 | 59 | GABA-B-R2 | 14 of 15 | ||
| NMDA receptor | GRIN2B | 27 | Nmdar2 | 12 of 15 | ||
| GRIN2D | 46 | 14 of 15 | ||||
| AMPA receptor | FRRS1L | 37 | - | - | ||
| Solute carrier family | SLC1A2 (EAAT2) | 41 | Eaat2 | 12 of 15 | ||
| SLC12A5 | 34 | kcc | 12 of 15 | |||
| SLC13A5 | 25 | Indy | 12 of 15 | |||
| SLC25A22 | 3 | GC1 | 15 of 15 | |||
| SLC25A12 | 39 | aralar1 | 15 of 15 | |||
| SLC35A2 | 22 | Ugalt | 13 of 15 | |||
| Synaptic vesicle release/membrane trafficking | DNM1 | 31 | shi | 14 of 15 | ||
| STXBP1 | 4 | Rop | 15 of 15 | |||
| CPLX1 | 63 | cpx | 7 of 15 | |||
| NECAP1 | 21 | CG9132 | 13 of 15 | |||
| TRAK1 | 68 | milt | 13 of 15 | |||
| AP3B2 | 48 | rb | 10 of 15 | |||
| SYNJ1 | 53 | Synj | 13 of 15 | |||
| TBC1D24 | 16 | sky | 15 of 15 | |||
| Cell adhision molecule | PCDH19 | 9 | - | - | ||
| ADAM22 | 61 | mmd | 8 of 15 | |||
| Cytoskeletal protein | SPTAN1 | 5 | α-Spec | 15 of 15 | ||
| PHACTR1 | 70 | CG32264 | 6 of 15 | |||
| Intracellular signal transduction | GNAO1 | 17 | Gαo | 15 of 15 | ||
| CDKL5 | 2 | - | - | |||
| RHOBTB2 | 64 | RhoBTB | 13 of 15 | |||
| PLCB1 | 12 | Plc21C | 14 of 15 | |||
| FGF12 | 47 | - | - | |||
| SIK1 | 30 | - | - | |||
| ARHGEF9 | 8 | - | - | |||
| DOCK7 | 23 | Zir | 15 of 15 | |||
| DENND5A | 49 | pns | 14 of 15 | |||
| SZT2 | 18 | - | - | |||
| YWHAG | 56 | 14-3-3ε | 15 of 15 | |||
| NTRK2 | 58 | - | - | |||
| CYFIP2 | 65 | Sra-1 | 15 of 15 | |||
| DMXL2 | 81 | Rbcn-3A | 11 of 15 | |||
| Transcription factor | ARX | 1 | al | 6 of 15 | ||
| CUX2 | 67 | ct | 8 of 15 | |||
| NEUROD2 | 72 | - | - | |||
| Translation | EEF1A2 | 33 | eEF1α2 | 12 of 15 | ||
| AARS | 29 | AlaRS | 14 of 15 | |||
| PARS2 | 75 | ProRS-m | 13 of 15 | |||
| GUF1 | 40 | waw | 13 of 15 | |||
| Post-translational modification | UBA5 | 44 | Uba5 | 15 of 15 | ||
| RNF13 | 73 | gzl | 10 of 15 | |||
| ALG13 | 36 | otu, CG14512 | 8 of 15 | |||
| PIGQ | 77 | PIG-Q | 6 of 15 | |||
| PIGP | 55 | PIG-P | 9 of 15 | |||
| PIGB | 80 | PIG-B | 13 of 15 | |||
| PIGA | 20 | PIG-A | 15 of 15 | |||
| Epigenetic factor | ACTL6B | 76 | Bap55 | 14 of 15 | ||
| Mitochondrial enzyme | MDH2 | 51 | Mdh2 | 14 of 15 | ||
| GLS | 71 | GLS | 14 of 15 | |||
| GOT2 | 82 | Got2 | 14 of 15 | |||
| Others | PNKP | 10 | CG9601 | 13 of 15 | ||
| ST3GAL3 | 15 | - | - | |||
| CAD | 50 | r | 14 of 15 | |||
| ITPA | 35 | CG8891 | 15 of 15 | |||
| HNRNPU | 54 | CG30122 | 11 of 15 | |||
| WWOX | 28 | Wwox | 13 of 15 | |||
| ARV1 | 38 | Arv1 | 11 of 15 | |||
| CNPY3 | 60 | CNPYb | 10 of 15 | |||
| PACS2 | 66 | KrT95D | 13 of 15 | |||
| UGP2 | 83 | UGP | 12 of 15 | |||
| UGDH | 84 | sgl | 15 of 15 | |||
| Species | Drosophila melanogaster | Caenorhabditis elegans | Danio rerio (Zebrafish) | Mus musculus (Mice) | iPSC (Human) |
|---|---|---|---|---|---|
| Number of neurons | 135,000 | 302 | ~1,000,000 | 71,000,000 | - |
| Genome size | 0.14 Gbp | 0.10 Gbp | 1.4 Gbp | 2.8 Gbp | 6.3 Gbp |
| Human disease genes conservative rate | 75% | 65% | 82% | 99% | 100% |
| Generation time | 10 days | 4 days | 3–4 months * | 9–11 weeks | - |
| Complex behavior | + | + | ++ | +++ | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takai, A.; Yamaguchi, M.; Yoshida, H.; Chiyonobu, T. Investigating Developmental and Epileptic Encephalopathy Using Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 6442. https://doi.org/10.3390/ijms21176442
Takai A, Yamaguchi M, Yoshida H, Chiyonobu T. Investigating Developmental and Epileptic Encephalopathy Using Drosophila melanogaster. International Journal of Molecular Sciences. 2020; 21(17):6442. https://doi.org/10.3390/ijms21176442
Chicago/Turabian StyleTakai, Akari, Masamitsu Yamaguchi, Hideki Yoshida, and Tomohiro Chiyonobu. 2020. "Investigating Developmental and Epileptic Encephalopathy Using Drosophila melanogaster" International Journal of Molecular Sciences 21, no. 17: 6442. https://doi.org/10.3390/ijms21176442
APA StyleTakai, A., Yamaguchi, M., Yoshida, H., & Chiyonobu, T. (2020). Investigating Developmental and Epileptic Encephalopathy Using Drosophila melanogaster. International Journal of Molecular Sciences, 21(17), 6442. https://doi.org/10.3390/ijms21176442