Are Alterations in Skeletal Muscle Mitochondria a Cause or Consequence of Insulin Resistance?


Abstract
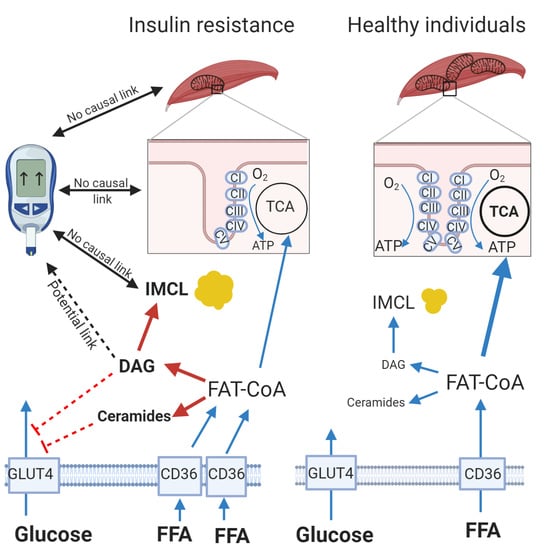
:
1. Introduction
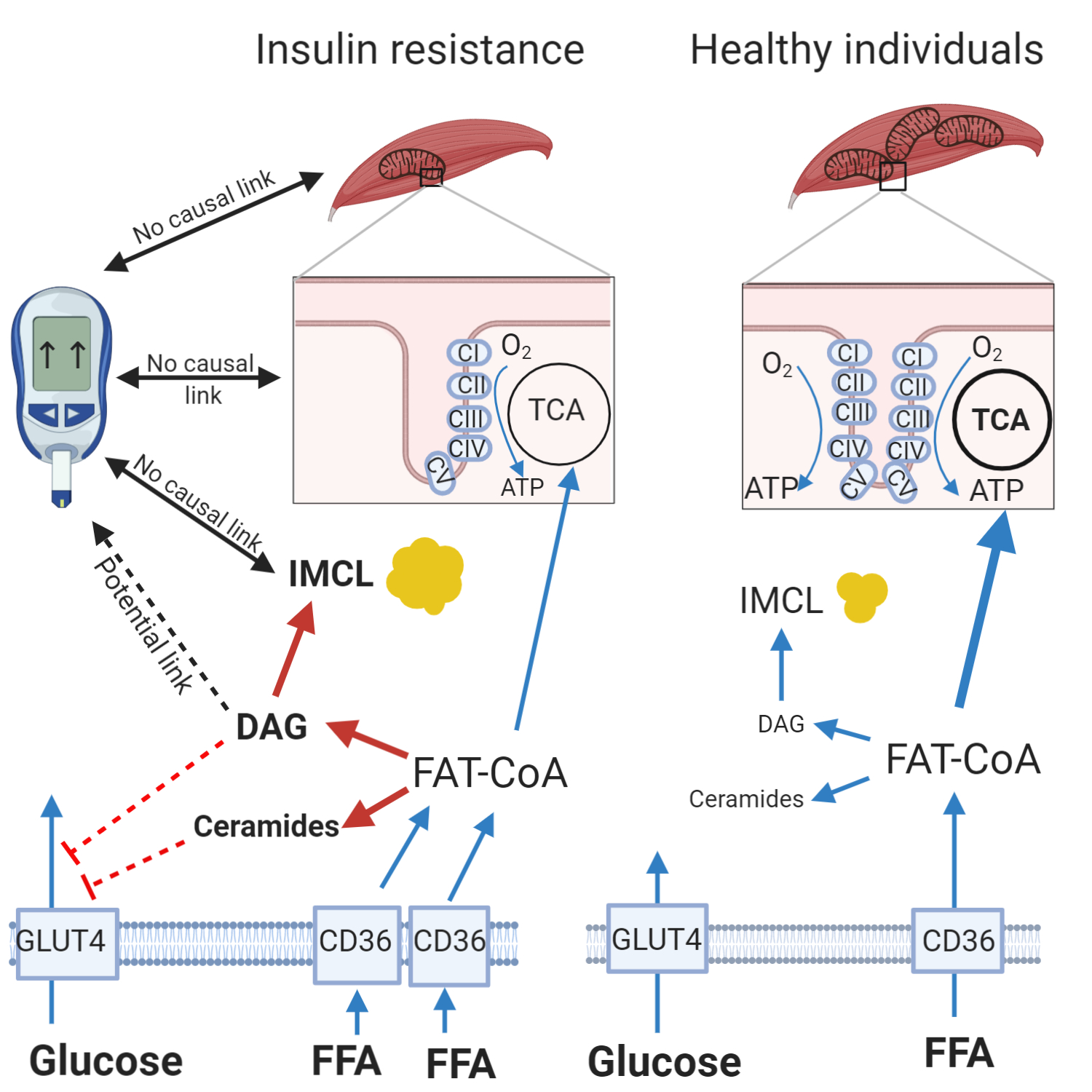
2. Mitochondria
Mitochondrial Terminology
3. Insulin Resistance
4. Mitochondrial Content and Insulin Resistance
4.1. Methods of Mitochondrial Content Measurement
4.2. Mitochondrial Content in Patients With Insulin Resistance or Type 2 Diabetes
4.3. Correlations between Mitochondrial Content and Insulin Resistance
4.4. Interventions That Alter Mitochondrial Content and Their Effects on Insulin Resistance
4.5. Experimental Animal Models
4.6. Conclusions
5. Mitochondrial Respiratory Function
5.1. Mitochondrial Respiratory Function in Patients with Insulin Resistance or Type 2 Diabetes
5.2. Is There a Relationship between Mitochondrial Respiratory Function and Insulin Resistance?
5.3. Interventions That Alter Mitochondrial Respiration and Their Effects on Insulin Resistance
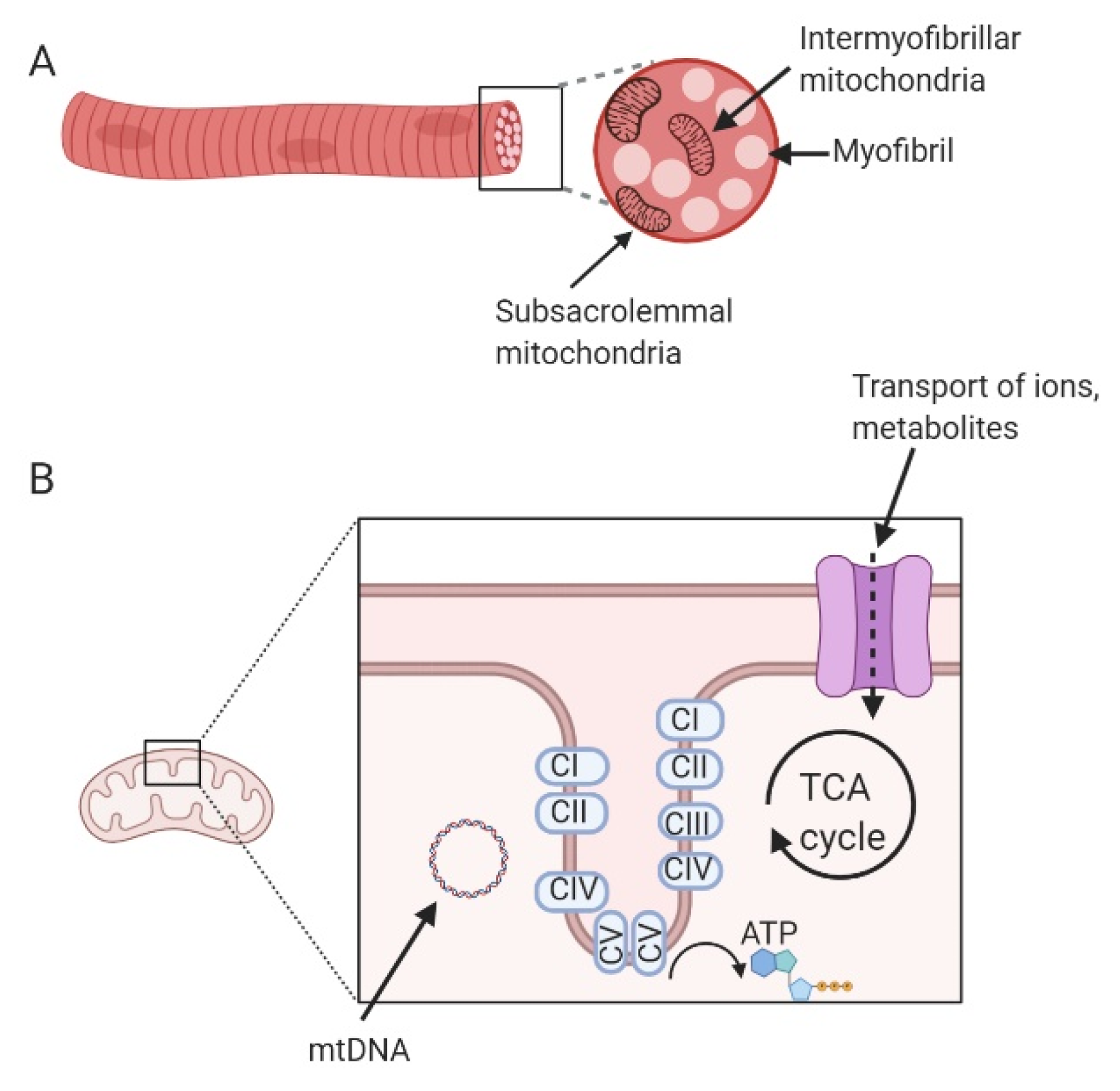
5.3.1. Physical Activity
5.3.2. Weight Loss
5.4. Experimental Animal Models of Insulin Resistance and Mitochondrial Respiration
5.5. Summary
6. Fatty Acid Metabolism and Insulin Resistance
6.1. Increased Inter- and Intramyocellular Lipids in Patients with Obesity and Type 2 Diabetes
6.2. Evidence for a Role of Impaired Mitochondrial Fatty Acid Metabolism in the Development of Insulin Resistance
6.3. The Influence of Exercise Training and Weight Loss on Imcl Stores and Fatty Acid Oxidation
6.4. Evidence from Animal Models
6.5. Summary
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Β-HAD | Beta-hydroxyacyl CoA dehydrogenase |
| BMI | Body mass index |
| CPT1 | Carnitine palmitoyltransferase |
| CS | Citrate synthase |
| CI-CV | Complexes I–V of the electron transport chain |
| DAG | Diacylglycerol |
| ETC | Electron transport chain |
| FAO | Fatty acid oxidation |
| GK | Goto-Kakizaki (rats) |
| HbA1c | Glycated haemoglobin |
| HFD | High fat diet |
| HOMA | Homeostatic model assessment |
| HSP | Heat shock protein |
| IMF | Intermyofibrillar |
| IMCL | Intramyocellular lipid |
| IP | Intraperitoneal |
| MAPR | Mitochondrial ATP production rate |
| mtDNA | Mitochondrial DNA |
| OXPHOS | Oxidative phosphorylation |
| PGC-1α | Peroxisome proliferator-activated receptor gamma co-activator 1alpha |
| 31P MRS | Phosphorus magnetic resonance spectroscopy |
| ROS | Reactive oxygen species |
| SS | Subsarcolemmal |
| TCA | Tricarboxylic acid |
| TEM | Transmission electron microscopy |
| TG | Triglyceride |
| T2D | Type 2 diabetes |
| UCP3 | Uncoupling protein 3 |
| ZDF | Zucker diabetic fatty (rat) |
References
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Thiebaud, D.; Jacot, E.; DeFronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.P. The effect of graded doses of insulin on total glucose uptake, glucose oxidation, and glucose storage in man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar]
- Stanford, K.I.; Goodyear, L.J. Exercise and type 2 diabetes: Molecular mechanisms regulating glucose uptake in skeletal muscle. Adv. Physiol. Educ. 2014, 38, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Meex, R.C.; Schrauwen-Hinderling, V.B.; Moonen-Kornips, E.; Schaart, G.; Mensink, M.; Phielix, E.; van de Weijer, T.; Sels, J.P.; Schrauwen, P.; Hesselink, M.K. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes 2010, 59, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Meex, R.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia 2010, 53, 1714–1721. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.; Bowen, B.P.; Lefort, N.; Flynn, C.R.; De Filippis, E.A.; Roberts, C.; Smoke, C.C.; Meyer, C.; Hojlund, K.; Yi, Z.; et al. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes 2010, 59, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.J.; Botella, J.; Genders, A.J.; Lee, M.J.; Saner, N.J.; Kuang, J.; Yan, X.; Granata, C. High-Intensity Exercise and Mitochondrial Biogenesis: Current Controversies and Future Research Directions. Physiology 2019, 34, 56–70. [Google Scholar] [CrossRef]
- Granata, C.; Oliveira, R.S.; Little, J.P.; Renner, K.; Bishop, D.J. Training intensity modulates changes in PGC-1alpha and p53 protein content and mitochondrial respiration, but not markers of mitochondrial content in human skeletal muscle. FASEB J. 2016, 30, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar]
- Hordern, M.D.; Dunstan, D.W.; Prins, J.B.; Baker, M.K.; Singh, M.A.; Coombes, J.S. Exercise prescription for patients with type 2 diabetes and pre-diabetes: A position statement from Exercise and Sport Science Australia. J. Sci. Med. Sport 2012, 15, 25–31. [Google Scholar] [CrossRef]
- Colberg, S.R.; Sigal, R.J.; Yardley, J.E.; Riddell, M.C.; Dunstan, D.W.; Dempsey, P.C.; Horton, E.S.; Castorino, K.; Tate, D.F. Physical Activity/Exercise and Diabetes: A Position Statement of the American Diabetes Association. Diabetes Care 2016, 39, 2065–2079. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Toledo, F.G.; Goodpaster, B.H. The role of weight loss and exercise in correcting skeletal muscle mitochondrial abnormalities in obesity, diabetes and aging. Mol. Cell Endocrinol. 2013, 379, 30–34. [Google Scholar] [CrossRef]
- Hood, D.A. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. 2001, 90, 1137–1157. [Google Scholar] [CrossRef]
- Yan, Z.; Lira, V.A.; Greene, N.P. Exercise training-induced regulation of mitochondrial quality. Exerc. Sport Sci. Rev. 2012, 40, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Koves, T.R.; Noland, R.C.; Bates, A.L.; Henes, S.T.; Muoio, D.M.; Cortright, R.N. Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am. J. Physiol. Cell Physiol. 2005, 288, C1074–C1082. [Google Scholar] [CrossRef] [Green Version]
- Bizeau, M.E.; Willis, W.T.; Hazel, J.R. Differential responses to endurance training in subsarcolemmal and intermyofibrillar mitochondria. J. Appl. Physiol. 1998, 85, 1279–1284. [Google Scholar] [CrossRef]
- Short, K.R. Measuring mitochondrial protein synthesis to assess biogenesis. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1153–E1154. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.F.; Hamilton, K.L. A perspective on the determination of mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E496–E499. [Google Scholar] [CrossRef] [Green Version]
- Atherton, P.J.; Phillips, B.E.; Wilkinson, D.J. Exercise and Regulation of Protein Metabolism. Prog. Mol. Biol. Transl. Sci. 2015, 135, 75–98. [Google Scholar] [CrossRef]
- Bishop, D.J.; Granata, C.; Eynon, N. Can we optimise the exercise training prescription to maximise improvements in mitochondria function and content? Biochim. Biophys. Acta 2014, 1840, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Lebon, V.; Dufour, S.; Petersen, K.F.; Ren, J.; Jucker, B.M.; Slezak, L.A.; Cline, G.W.; Rothman, D.L.; Shulman, G.I. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J. Clin. Investig. 2001, 108, 733–737. [Google Scholar] [CrossRef]
- Lanza, I.R.; Nair, K.S. Mitochondrial metabolic function assessed in vivo and in vitro. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Miotto, P.M.; LeBlanc, P.J.; Holloway, G.P. High-Fat Diet Causes Mitochondrial Dysfunction as a Result of Impaired ADP Sensitivity. Diabetes 2018, 67, 2199–2205. [Google Scholar] [CrossRef] [Green Version]
- Asmann, Y.W.; Stump, C.S.; Short, K.R.; Coenen-Schimke, J.M.; Guo, Z.; Bigelow, M.L.; Nair, K.S. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 2006, 55, 3309–3319. [Google Scholar] [CrossRef] [Green Version]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef]
- Ayala, J.E.; Samuel, V.T.; Morton, G.J.; Obici, S.; Croniger, C.M.; Shulman, G.I.; Wasserman, D.H.; McGuinness, O.P. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Model. Mech. 2010, 3, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; DeFronzo, R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care 1999, 22, 1462–1470. [Google Scholar] [CrossRef]
- Chomentowski, P.; Coen, P.M.; Radikova, Z.; Goodpaster, B.H.; Toledo, F.G. Skeletal muscle mitochondria in insulin resistance: Differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J. Clin. Endocrinol. Metab. 2011, 96, 494–503. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1039–E1044. [Google Scholar] [CrossRef] [Green Version]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsoe, R.; Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Van Tienen, F.H.; Praet, S.F.; de Feyter, H.M.; van den Broek, N.M.; Lindsey, P.J.; Schoonderwoerd, K.G.; de Coo, I.F.; Nicolay, K.; Prompers, J.J.; Smeets, H.J.; et al. Physical activity is the key determinant of skeletal muscle mitochondrial function in type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3261–3269. [Google Scholar] [CrossRef]
- Holloway, G.P.; Thrush, A.B.; Heigenhauser, G.J.; Tandon, N.N.; Dyck, D.J.; Bonen, A.; Spriet, L.L. Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1782–E1789. [Google Scholar] [CrossRef] [Green Version]
- Goodpaster, B.H. Mitochondrial deficiency is associated with insulin resistance. Diabetes 2013, 62, 1032–1035. [Google Scholar] [CrossRef] [Green Version]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Bruce, C.R.; Mertz, V.A.; Heigenhauser, G.J.; Dyck, D.J. The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes 2005, 54, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Nair, K.S.; Bigelow, M.L.; Asmann, Y.W.; Chow, L.S.; Coenen-Schimke, J.M.; Klaus, K.A.; Guo, Z.K.; Sreekumar, R.; Irving, B.A. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes 2008, 57, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.; Mogensen, M.; Vind, B.F.; Sahlin, K.; Højlund, K.; Schrøder, H.D.; Ortenblad, N. Increased subsarcolemmal lipids in type 2 diabetes: Effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E706–E713. [Google Scholar] [CrossRef] [Green Version]
- Ukropcova, B.; Sereda, O.; de Jonge, L.; Bogacka, I.; Nguyen, T.; Xie, H.; Bray, G.A.; Smith, S.R. Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes 2007, 56, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Toledo, F.G.; Menshikova, E.V.; Ritov, V.B.; Azuma, K.; Radikova, Z.; DeLany, J.; Kelley, D.E. Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes 2007, 56, 2142–2147. [Google Scholar] [CrossRef] [Green Version]
- Granata, C.; Jamnick, N.A.; Bishop, D.J. Training-Induced Changes in Mitochondrial Content and Respiratory Function in Human Skeletal Muscle. Sports Med. 2018, 48, 1809–1828. [Google Scholar] [CrossRef]
- Bordenave, S.; Metz, L.; Flavier, S.; Lambert, K.; Ghanassia, E.; Dupuy, A.M.; Michel, F.; Puech-Cathala, A.M.; Raynaud, E.; Brun, J.F.; et al. Training-induced improvement in lipid oxidation in type 2 diabetes mellitus is related to alterations in muscle mitochondrial activity. Effect of endurance training in type 2 diabetes. Diabetes Metab. 2008, 34, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Konopka, A.R.; Asante, A.; Lanza, I.R.; Robinson, M.M.; Johnson, M.L.; Man, C.D.; Cobelli, C.; Amols, M.H.; Irving, B.A.; Nair, K.S. Defects in mitochondrial efficiency and H2O2 emissions in obese women are restored to a lean phenotype with aerobic exercise training. Diabetes 2015, 64, 2104–2115. [Google Scholar] [CrossRef] [Green Version]
- Menshikova, E.V.; Ritov, V.B.; Toledo, F.G.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E818–E825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, C.R.; Thrush, A.B.; Mertz, V.A.; Bezaire, V.; Chabowski, A.; Heigenhauser, G.J.; Dyck, D.J. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E99–E107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, R.; Nederveen, J.P.; Gillen, J.B.; Joanisse, S.; Parise, G.; Tarnopolsky, M.A.; Gibala, M.J. Skeletal muscle fiber-type-specific changes in markers of capillary and mitochondrial content after low-volume interval training in overweight women. Physiol. Rep. 2018, 6, e13597. [Google Scholar] [CrossRef]
- Kenny, H.C.; Rudwill, F.; Breen, L.; Salanova, M.; Blottner, D.; Heise, T.; Heer, M.; Blanc, S.; O’Gorman, D.J. Bed rest and resistive vibration exercise unveil novel links between skeletal muscle mitochondrial function and insulin resistance. Diabetologia 2017, 60, 1491–1501. [Google Scholar] [CrossRef]
- Dirks, M.L.; Miotto, P.M.; Goossens, G.H.; Senden, J.M.; Petrick, H.L.; van Kranenburg, J.; van Loon, L.J.C.; Holloway, G.P. Short-term bed rest-induced insulin resistance cannot be explained by increased mitochondrial H(2) O(2) emission. J. Physiol. 2020, 598, 123–137. [Google Scholar] [CrossRef]
- Hey-Mogensen, M.; Hojlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernstrom, M.; Sahlin, K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, C.L.; Fealy, C.E.; Mulya, A.; Kirwan, J.P. Exercise Training Remodels Human Skeletal Muscle Mitochondrial Fission and Fusion Machinery Towards a Pro-Elongation Phenotype. Acta Physiol. 2018, 225, e13216. [Google Scholar] [CrossRef]
- Samjoo, I.A.; Safdar, A.; Hamadeh, M.J.; Glover, A.W.; Mocellin, N.J.; Santana, J.; Little, J.P.; Steinberg, G.R.; Raha, S.; Tarnopolsky, M.A. Markers of skeletal muscle mitochondrial function and lipid accumulation are moderately associated with the homeostasis model assessment index of insulin resistance in obese men. PLoS ONE 2013, 8, e66322. [Google Scholar] [CrossRef]
- Toledo, F.G.; Menshikova, E.V.; Azuma, K.; Radikova, Z.; Kelley, C.A.; Ritov, V.B.; Kelley, D.E. Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes 2008, 57, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Little, J.P.; Gillen, J.B.; Percival, M.E.; Safdar, A.; Tarnopolsky, M.A.; Punthakee, Z.; Jung, M.E.; Gibala, M.J. Low-volume high-intensity interval training reduces hyperglycemia and increases muscle mitochondrial capacity in patients with type 2 diabetes. J. Appl. Physiol. 2011, 111, 1554–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogensen, M.; Vind, B.F.; Hojlund, K.; Beck-Nielsen, H.; Sahlin, K. Maximal lipid oxidation in patients with type 2 diabetes is normal and shows an adequate increase in response to aerobic training. Diabetes Obes. Metab. 2009, 11, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Schleh, M.W.; Ahn, C.; Ludzki, A.C.; Gillen, J.B.; Varshney, P.; van Pelt, D.W.; Pitchford, L.M.; Chenevert, T.L.; Gioscia-Ryan, R.A.; et al. Moderate-intensity exercise and high-intensity interval training affect insulin sensitivity similarly in obese adults. J. Clin. Endocrinol. Metab. 2020, 105, dgaa345. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Rizza, R.A.; Coenen-Schimke, J.M.; Nair, K.S. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 2003, 52, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Hood, M.S.; Little, J.P.; Tarnopolsky, M.A.; Myslik, F.; Gibala, M.J. Low-volume interval training improves muscle oxidative capacity in sedentary adults. Med. Sci. Sport Exerc. 2011, 43, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Irving, B.A.; Short, K.R.; Nair, K.S.; Stump, C.S. Nine days of intensive exercise training improves mitochondrial function but not insulin action in adult offspring of mothers with type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, E1137–E1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, S.K.; Teede, H.J.; Rachoń, D.; Harrison, C.L.; Strauss, B.J.; Stepto, N.K. Effect of exercise training on insulin sensitivity, mitochondria and computed tomography muscle attenuation in overweight women with and without polycystic ovary syndrome. Diabetologia 2012, 55, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Coen, P.M.; Menshikova, E.V.; Distefano, G.; Zheng, D.; Tanner, C.J.; Standley, R.A.; Helbling, N.L.; Dubis, G.S.; Ritov, V.B.; Xie, H.; et al. Exercise and Weight Loss Improve Muscle Mitochondrial Respiration, Lipid Partitioning, and Insulin Sensitivity After Gastric Bypass Surgery. Diabetes 2015, 64, 3737–3750. [Google Scholar] [CrossRef] [Green Version]
- Kras, K.A.; Hoffman, N.; Roust, L.R.; Benjamin, T.R.; EA, D.E.F.; Katsanos, C.S. Adenosine Triphosphate Production of Muscle Mitochondria after Acute Exercise in Lean and Obese Humans. Med. Sci. Sport Exerc. 2019, 51, 445–453. [Google Scholar] [CrossRef]
- Benton, C.R.; Holloway, G.P.; Han, X.X.; Yoshida, Y.; Snook, L.A.; Lally, J.; Glatz, J.F.; Luiken, J.J.; Chabowski, A.; Bonen, A. Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia 2010, 53, 2008–2019. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Skeletal muscle energetics are compromised only during high-intensity contractions in the Goto-Kakizaki rat model of type 2 diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R356–R368. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.P.; Gurd, B.J.; Snook, L.A.; Lally, J.; Bonen, A. Compensatory increases in nuclear PGC1alpha protein are primarily associated with subsarcolemmal mitochondrial adaptations in ZDF rats. Diabetes 2010, 59, 819–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lally, J.S.; Snook, L.A.; Han, X.X.; Chabowski, A.; Bonen, A.; Holloway, G.P. Subcellular lipid droplet distribution in red and white muscles in the obese Zucker rat. Diabetologia 2012, 55, 479–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.C.; Mullen, K.L.; Junkin, K.A.; Nickerson, J.; Chabowski, A.; Bonen, A.; Dyck, D.J. Metformin and exercise reduce muscle FAT/CD36 and lipid accumulation and blunt the progression of high-fat diet-induced hyperglycemia. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E172–E181. [Google Scholar] [CrossRef] [Green Version]
- Barbosa de Queiroz, K.; Honorato-Sampaio, K.; Rossoni Junior, J.V.; Andrade Leal, D.; Pinto, A.B.; Kappes-Becker, L.; Evangelista, E.A.; Guerra-Sa, R. Physical activity prevents alterations in mitochondrial ultrastructure and glucometabolic parameters in a high-sugar diet model. PLoS ONE 2017, 12, e0172103. [Google Scholar] [CrossRef] [Green Version]
- Buettner, R.; Parhofer, K.G.; Woenckhaus, M.; Wrede, C.E.; Kunz-Schughart, L.A.; Scholmerich, J.; Bollheimer, L.C. Defining high-fat-diet rat models: Metabolic and molecular effects of different fat types. J. Mol. Endocrinol. 2006, 36, 485–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Perez, Y.; Capllonch-Amer, G.; Gianotti, M.; Llado, I.; Proenza, A.M. Long-term high-fat-diet feeding induces skeletal muscle mitochondrial biogenesis in rats in a sex-dependent and muscle-type specific manner. Nutr. Metab. 2012, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Hancock, C.R.; Han, D.H.; Chen, M.; Terada, S.; Yasuda, T.; Wright, D.C.; Holloszy, J.O. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 7815–7820. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Bruce, C.R.; Beale, S.M.; Hoehn, K.L.; So, T.; Rolph, M.S.; Cooney, G.J. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: Evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 2007, 56, 2085–2092. [Google Scholar] [CrossRef] [Green Version]
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.S.; et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015, 58, 2381–2391. [Google Scholar] [CrossRef] [Green Version]
- Leduc-Gaudet, J.P.; Reynaud, O.; Chabot, F.; Mercier, J.; Andrich, D.E.; St-Pierre, D.H.; Gouspillou, G. The impact of a short-term high-fat diet on mitochondrial respiration, reactive oxygen species production, and dynamics in oxidative and glycolytic skeletal muscles of young rats. Physiol. Rep. 2018, 6, e13548. [Google Scholar] [CrossRef] [PubMed]
- Brunetta, H.S.; de Paula, G.C.; de Oliveira, J.; Martins, E.L.; Dos Santos, G.J.; Galina, A.; Rafacho, A.; de Bem, A.F.; Nunes, E.A. Decrement in resting and insulin-stimulated soleus muscle mitochondrial respiration is an early event in diet-induced obesity in mice. Exp. Physiol. 2019, 104, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. Skeletal muscle “mitochondrial deficiency” does not mediate insulin resistance. Am. J. Clin. Nutr. 2009, 89, 463s–466s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putti, R.; Migliaccio, V.; Sica, R.; Lionetti, L. Skeletal Muscle Mitochondrial Bioenergetics and Morphology in High Fat Diet Induced Obesity and Insulin Resistance: Focus on Dietary Fat Source. Front. Physiol. 2016, 20, 426. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.S.; Paglialunga, S.; Vigna, C.; Ludzki, A.; Herbst, E.A.; Lally, J.S.; Schrauwen, P.; Hoeks, J.; Tupling, A.R.; Bonen, A. High-fat diet-induced mitochondrial biogenesis is regulated by mitochondrial-derived reactive oxygen species activation of CaMKII. Diabetes 2014, 63, 1907–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, T.M.; Garcia-Martin, R.; Cai, W.; Konishi, M.; O’Neill, B.T.; Sakaguchi, M.; Kim, J.H.; Jung, D.Y.; Kim, J.K.; Kahn, C.R. Multi-dimensional Transcriptional Remodeling by Physiological Insulin In Vivo. Cell Rep. 2019, 26, 3429–3443. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Quantification of mitochondrial oxidative phosphorylation in metabolic disease: Application to type 2 diabetes. Int. J. Mol. Sci. 2019, 20, 5271. [Google Scholar] [CrossRef] [Green Version]
- Hesselink, M.K.; Schrauwen-Hinderling, V.; Schrauwen, P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 633–645. [Google Scholar] [CrossRef]
- Holloszy, J.O. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 2013, 62, 1036–1040. [Google Scholar] [CrossRef] [Green Version]
- Wredenberg, A.; Freyer, C.; Sandstrom, M.E.; Katz, A.; Wibom, R.; Westerblad, H.; Larsson, N.G. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem. Biophys. Res. Commun. 2006, 350, 202–207. [Google Scholar] [CrossRef]
- Han, D.H.; Nolte, L.A.; Ju, J.S.; Coleman, T.; Holloszy, J.O.; Semenkovich, C.F. UCP-mediated energy depletion in skeletal muscle increases glucose transport despite lipid accumulation and mitochondrial dysfunction. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E347–E353. [Google Scholar] [CrossRef] [PubMed]
- Cree-Green, M.; Gupta, A.; Coe, G.V.; Baumgartner, A.D.; Pyle, L.; Reusch, J.E.; Brown, M.S.; Newcomer, B.R.; Nadeau, K.J. Insulin resistance in type 2 diabetes youth relates to serum free fatty acids and muscle mitochondrial dysfunction. J. Diabetes Complicat. 2017, 31, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, I.R.; Nair, K.S. Muscle mitochondrial changes with aging and exercise. Am. J. Clin. Nutr. 2009, 89, 467s–471s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, E.; Chia, C.W.; Spencer, R.G.; Fishbein, K.W.; Reiter, D.A.; Cameron, D.; Zane, A.C.; Moore, Z.A.; Gonzalez-Freire, M.; Zoli, M.; et al. Insulin Resistance Is Associated With Reduced Mitochondrial Oxidative Capacity Measured by 31P-Magnetic Resonance Spectroscopy in Participants Without Diabetes From the Baltimore Longitudinal Study of Aging. Diabetes 2017, 66, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Tran, L.; Langlais, P.R.; Hoffman, N.; Roust, L.; Katsanos, C.S. Mitochondrial ATP synthase beta-subunit production rate and ATP synthase specific activity are reduced in skeletal muscle of humans with obesity. Exp. Physiol. 2019, 104, 126–135. [Google Scholar] [CrossRef]
- De Feyter, H.M.; van Den Broek, N.M.; Praet, S.F.; Nicolay, K.; van Loon, L.J.; Prompers, J.J. Early or advanced stage type 2 diabetes is not accompanied by in vivo skeletal muscle mitochondrial dysfunction. Eur. J. Endocrinol. 2008, 158, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Wellman, K.H.; Weber, T.M.; Cathey, B.L.; Brophy, P.M.; Gilliam, L.A.; Kane, C.L.; Maples, J.M.; Gavin, T.P.; Houmard, J.A.; Neufer, P.D. Mitochondrial respiratory capacity and content are normal in young insulin-resistant obese humans. Diabetes 2014, 63, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayer, A.A.; Dennison, E.M.; Syddall, H.E.; Gilbody, H.J.; Phillips, D.I.; Cooper, C. Type 2 diabetes, muscle strength, and impaired physical function: The tip of the iceberg? Diabetes Care 2005, 28, 2541–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, R.; Hojlund, K. Proteomic study of skeletal muscle in obesity and type 2 diabetes: Progress and potential. Expert Rev. Proteomics 2018, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Miotto, P.M.; McGlory, C.; Bahniwal, R.; Kamal, M.; Phillips, S.M.; Holloway, G.P. Supplementation with dietary ω-3 mitigates immobilization-induced reductions in skeletal muscle mitochondrial respiration in young women. FASEB J. 2019, 33, 8232–8240. [Google Scholar] [CrossRef] [PubMed]
- Rabol, R.; Svendsen, P.F.; Skovbro, M.; Boushel, R.; Haugaard, S.B.; Schjerling, P.; Schrauwen, P.; Hesselink, M.K.; Nilas, L.; Madsbad, S.; et al. Reduced skeletal muscle mitochondrial respiration and improved glucose metabolism in nondiabetic obese women during a very low calorie dietary intervention leading to rapid weight loss. Metabolism 2009, 58, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Scheede-Bergdahl, C.; Whitesell, T.; Boushel, R.; Bergdahl, A. Increased intrinsic mitochondrial respiratory capacity in skeletal muscle from rats with streptozotocin-induced hyperglycemia. Physiol. Rep. 2015, 3, e12467. [Google Scholar] [CrossRef]
- Brunmair, B.; Staniek, K.; Gras, F.; Scharf, N.; Althaym, A.; Clara, R.; Roden, M.; Gnaiger, E.; Nohl, H.; Waldhausl, W.; et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: A common mechanism contributing to their antidiabetic actions? Diabetes 2004, 53, 1052–1059. [Google Scholar] [CrossRef] [Green Version]
- Henstridge, D.C.; Bruce, C.R.; Drew, B.G.; Tory, K.; Kolonics, A.; Estevez, E.; Chung, J.; Watson, N.; Gardner, T.; Lee-Young, R.S.; et al. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 2014, 63, 1881–1894. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef] [Green Version]
- Tiganis, T. Reactive oxygen species and insulin resistance: The good, the bad and the ugly. Trends Pharmacol. Sci. 2011, 32, 82–89. [Google Scholar] [CrossRef]
- Formentini, L.; Ryan, A.J.; Gálvez-Santisteban, M.; Carter, L.; Taub, P.; Lapek, J.D., Jr.; Gonzalez, D.J.; Villarreal, F.; Ciaraldi, T.P.; Cuezva, J.M.; et al. Mitochondrial H(+)-ATP synthase in human skeletal muscle: Contribution to dyslipidaemia and insulin resistance. Diabetologia 2017, 60, 2052–2065. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Technol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr. Diabetes Rep. 2010, 10, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.; Rud, K.A.; Mortensen, O.H.; Frandsen, L.; Grunnet, N.; Quistorff, B. Your mitochondria are what you eat: A high-fat or a high-sucrose diet eliminates metabolic flexibility in isolated mitochondria from rat skeletal muscle. Physiol. Rep. 2017, 5, e13207. [Google Scholar] [CrossRef]
- Eldor, R.; Norton, L.; Fourcaudot, M.; Galindo, C.; DeFronzo, R.A.; Abdul-Ghani, M. Increased lipid availability for three days reduces whole body glucose uptake, impairs muscle mitochondrial function and initiates opposing effects on PGC-1alpha promoter methylation in healthy subjects. PLoS ONE 2017, 12, e0188208. [Google Scholar] [CrossRef] [Green Version]
- Dandanell, S.; Meinild-Lundby, A.K.; Andersen, A.B.; Lang, P.F.; Oberholzer, L.; Keiser, S.; Robach, P.; Larsen, S.; Ronnestad, B.R.; Lundby, C. Determinants of maximal whole-body fat oxidation in elite cross-country skiers: Role of skeletal muscle mitochondria. Scand. J. Med. Sci. Sports 2018, 28, 2494–2504. [Google Scholar] [CrossRef]
- Schenk, S.; Horowitz, J.F. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J. Clin. Investig. 2007, 117, 1690–1698. [Google Scholar] [CrossRef]
- Zacharewicz, E.; Hesselink, M.K.C.; Schrauwen, P. Exercise counteracts lipotoxicity by improving lipid turnover and lipid droplet quality. J. Intern. Med. 2018, 284, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Bergman, B.C.; Perreault, L.; Strauss, A.; Bacon, S.; Kerege, A.; Harrison, K.; Brozinick, J.T.; Hunerdosse, D.M.; Playdon, M.C.; Holmes, W.; et al. Intramuscular triglyceride synthesis: Importance in muscle lipid partitioning in humans. Am. J. Physiol. Endocrinol. Meta 2018, 314, e152–e164. [Google Scholar] [CrossRef]
- Kiens, B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol. Rev. 2006, 86, 205–243. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.L.; Hattersley, J.; Frost, G.S.; Chambers, E.S.; Wallis, G.A. Maximal fat oxidation during exercise is positively associated with 24-h fat oxidation and insulin sensitivity in young, healthy men. J. Appl. Physiol. 2015, 118, 1415–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lira, V.A.; Benton, C.R.; Yan, Z.; Bonen, A. PGC-1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E145–E161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanishi, N.; Takagi, K.; Lee, H.C.; Nakano, D.; Okuno, T.; Yokomizo, T.; Machida, S. Endurance exercise training and high-fat diet differentially affect composition of diacylglycerol molecular species in rat skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R892–R901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudz, T.I.; Tserng, K.Y.; Hoppel, C.L. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 1997, 272, 24154–24158. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef] [PubMed]
- Perreault, L.; Newsom, S.A.; Strauss, A.; Kerege, A.; Kahn, D.E.; Harrison, K.A.; Snell-Bergeon, J.K.; Nemkov, T.; D’Alessandro, A.; Jackman, M.R.; et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 2018, 3, e96805. [Google Scholar] [CrossRef] [Green Version]
- Coen, P.M.; Dube, J.J.; Amati, F.; Stefanovic-Racic, M.; Ferrell, R.E.; Toledo, F.G.; Goodpaster, B.H. Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes 2010, 59, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Toledo, F.G.S.; Johannsen, D.L.; Covington, J.D.; Bajpeyi, S.; Goodpaster, B.; Conley, K.E.; Ravussin, E. Impact of prolonged overfeeding on skeletal muscle mitochondria in healthy individuals. Diabetologia 2018, 61, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Brehm, A.; Krssak, M.; Schmid, A.I.; Nowotny, P.; Waldhausl, W.; Roden, M. Acute elevation of plasma lipids does not affect ATP synthesis in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E33–E38. [Google Scholar] [CrossRef] [Green Version]
- Gavin, T.P.; Ernst, J.M.; Kwak, H.B.; Caudill, S.E.; Reed, M.A.; Garner, R.T.; Nie, Y.; Weiss, J.A.; Pories, W.J.; Dar, M.; et al. High Incomplete Skeletal Muscle Fatty Acid Oxidation Explains Low Muscle Insulin Sensitivity in Poorly Controlled T2D. J. Clin. Endocrinol. Metab. 2018, 103, 882–889. [Google Scholar] [CrossRef]
- Ludzki, A.; Paglialunga, S.; Smith, B.K.; Herbst, E.A.; Allison, M.K.; Heigenhauser, G.J.; Neufer, P.D.; Holloway, G.P. Rapid Repression of ADP Transport by Palmitoyl-CoA Is Attenuated by Exercise Training in Humans: A Potential Mechanism to Decrease Oxidative Stress and Improve Skeletal Muscle Insulin Signaling. Diabetes 2015, 64, 2769–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, L.E.; Brandon, A.E.; Hoy, A.J.; Forsberg, G.B.; Lelliott, C.J.; Reznick, J.; Löfgren, L.; Oscarsson, J.; Strömstedt, M.; Cooney, G.J.; et al. Amelioration of lipid-induced insulin resistance in rat skeletal muscle by overexpression of Pgc-1β involves reductions in long-chain acyl-CoA levels and oxidative stress. Diabetologia 2011, 54, 1417–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicks, S.E.; Vandanmagsar, B.; Haynie, K.R.; Fuller, S.E.; Warfel, J.D.; Stephens, J.M.; Wang, M.; Han, X.; Zhang, J.; Noland, R.C.; et al. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, E3300–E3309. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Wicks, S.E.; Vandanmagsar, B.; Mendoza, T.M.; Bayless, D.S.; Salbaum, J.M.; Dearth, S.P.; Campagna, S.R.; Mynatt, R.L.; Noland, R.C. Extensive metabolic remodeling after limiting mitochondrial lipid burden is consistent with an improved metabolic health profile. J Biol Chem 2019, 294, 12313–12327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, C.R.; Hoy, A.J.; Turner, N.; Watt, M.J.; Allen, T.L.; Carpenter, K.; Cooney, G.J.; Febbraio, M.A.; Kraegen, E.W. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes 2009, 58, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Gemmink, A.; Schrauwen, P.; Hesselink, M.K.C. Exercising your fat (metabolism) into shape: A muscle-centred view. Diabetologia 2020, 63, 1453–1463. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Katsiaras, A.; Kelley, D.E. Enhanced fat oxidation through physical activity is associated with improvements in insulin sensitivity in obesity. Diabetes 2003, 52, 2191–2197. [Google Scholar] [CrossRef] [Green Version]
- Dube, J.J.; Amati, F.; Toledo, F.G.; Stefanovic-Racic, M.; Rossi, A.; Coen, P.; Goodpaster, B.H. Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 2011, 54, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Pino, M.F.; Stephens, N.A.; Eroshkin, A.M.; Yi, F.; Hodges, A.; Cornnell, H.H.; Pratley, R.E.; Smith, S.R.; Wang, M.; Han, X.; et al. Endurance training remodels skeletal muscle phospholipid composition and increases intrinsic mitochondrial respiration in men with Type 2 diabetes. Physiol. Genomics 2019, 51, 586–595. [Google Scholar] [CrossRef]
- Gonzalez-Armenta, J.L.; Gao, Z.; Appt, S.E.; Vitolins, M.Z.; Michalson, K.T.; Register, T.C.; Shively, C.A.; Molina, A.J.A. Skeletal Muscle Mitochondrial Respiration Is Elevated in Female Cynomolgus Macaques Fed a Western Compared with a Mediterranean Diet. J. Nutr 2019, 149, 1493–1502. [Google Scholar] [CrossRef]
- Hoehn, K.L.; Turner, N.; Swarbrick, M.M.; Wilks, D.; Preston, E.; Phua, Y.; Joshi, H.; Furler, S.M.; Larance, M.; Hegarty, B.D.; et al. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab. 2010, 11, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, D.P.; Pulinilkunnil, T.; Cline, G.W.; Shulman, G.I.; Lowell, B.B. Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake. Proc. Natl Acad Sci USA 2010, 107, 7598–7603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miotto, P.M.; Petrick, H.L.; Holloway, G.P. Acute insulin deprivation results in altered mitochondrial substrate sensitivity conducive to greater fatty acid transport. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E345–E353. [Google Scholar] [CrossRef]
- Schonke, M.; Bjornholm, M.; Chibalin, A.V.; Zierath, J.R.; Deshmukh, A.S. Proteomics Analysis of Skeletal Muscle from Leptin-Deficient ob/ob Mice Reveals Adaptive Remodeling of Metabolic Characteristics and Fiber Type Composition. Proteomics 2018, 18, e1700375. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
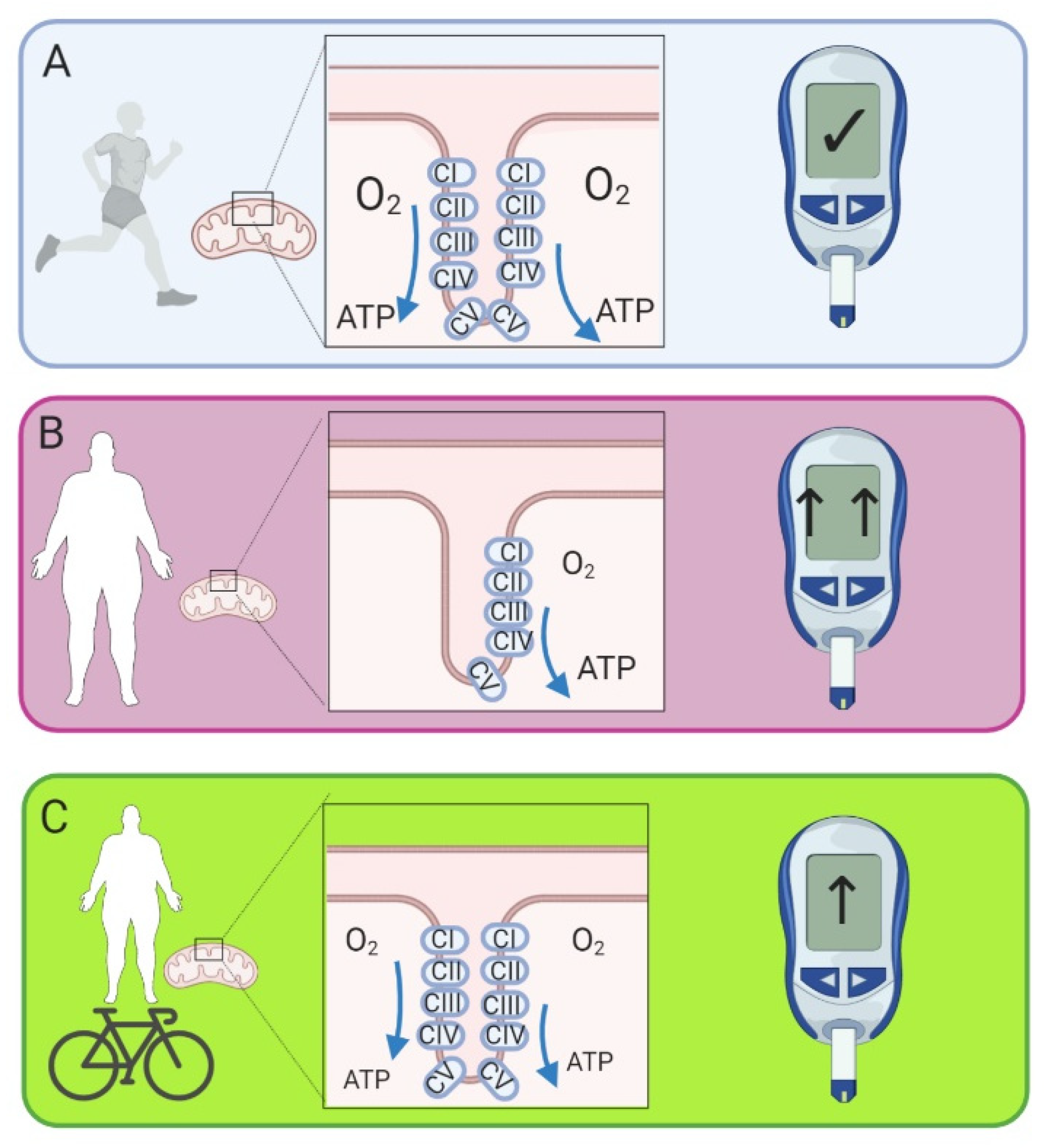{kind=link}
| Study | Population | Method | Finding |
|---|---|---|---|
| Chomentowski et al., 2011 [36] | T2D Non-diabetic IR | TEM | ↓ IMF, no change SS ↓ IMF, no change SS |
| Ritov et al., 2005 [7] | T2D | TEM | ↓ SS |
| Kelley et al., 2002 [5] | Obese T2D | CS | No change ↓ |
| Kim et al., 2000 [37] | Obese | CS | ↓ |
| Asmann et al., 2006 [32] | T2D | mtDNA | No change |
| Boushel et al., 2007 [38] | T2D | mtDNA | ↑ |
| Nair et al., 2008 [45] | T2D | CS | ↓ |
| van Tienen et al., 2012 [39] | T2D IR | CS | ↓ No change |
| Mogensen et al., 2007 [43] | T2D | CS | No change |
| Holloway et al., 2007 [40] | Obese | CS | ↓ |
| Bruce et al., 2005 [44] | Obese | CS | No change |
| Study | Participants | Exercise Training | Insulin Resistance Outcome | Mitochondria Outcome |
|---|---|---|---|---|
| Short et al., 2003 [64] | Male and female, young and older participants (21–87 y) Healthy, low regular activity level, normal weight | 16 weeks moderate-intensity exercise training | ↑ insulin sensitivity only in younger participants | ↑ mitochondrial gene expression ↑ mitochondrial enzyme activity |
| Menshikova et al., 2005 [52] | Male and female, overweight and obese, non-diabetic, sedentary | 16 weeks, 60–70% maximal intensity for 30–40 min for 4–6 sessions per week | ↑ insulin sensitivity | ↑ activity of ETC enzymes |
| Bruce et al., 2006 [53] | Male and female, obese, sedentary non-diabetic | 8 weeks, 5 days per week for 60 min at 65–70% of VO2 peak | ↑ glucose tolerance | ↑ fatty acid oxidation ↑ CPT1 activity ↑ β-HAD activity |
| Toledo et al., 2007 [48] | Sedentary, overweight/obese T2D | 16–20 weeks moderate intensity | ↑ insulin sensitivity | ↑ mitochondrial content ↑ mitochondrial enzymes |
| Meex et al., 2010 [8] | Male T2D and healthy controls, overweight and obese, sedentary | 12 weeks, 2 days per week For 30 min at 55% Wmax aerobic exercise plus one session of resistance exercise per week—8 reps at 55% MVC and 2 series of 8 reps at 75% MVC | ↑ insulin sensitivity | ↑ mitochondrial function (31P-MRS) |
| Phielix et al., 2010 [9] | As for Meex et al. | As for Meex et al. | ↑ insulin sensitivity | ↑ mitochondrial function (HRR) ↑ mtDNA |
| Bordenave et al., 2008 [50] | Male T2D, overweight, sedentary | 10 weeks, 2 days per week for 45 min at low-moderate intensity | No change in blood glucose | ↑ lipid oxidation ↑ respiration ↑ CS activity |
| Little et al., 2011 [61] | T2D patients, obese, mostly sedentary | 6 HIIT sessions over 2 weeks, 10 × 60 s intervals at 90% HRmax | ↓ hyperglycaemia ↑ GLUT4 | ↑ CS activity ↑ protein content of ETC complexes |
| Mogensen et al., 2009 [62] | Male T2D and controls, obese, similar activity levels in both groups (non-sedentary) | 10 weeks, 5 days per week for 30 min moderate intensity interval and continuous training | ↑ insulin sensitivity | ↑ CS activity post exercise training, but not different in T2D to controls |
| Hey-Mogensen et al., 2010 [57] | Male T2D and controls, obese, non-sedentary | 10 weeks, 4–5 days/week, moderate intensity | ↑ insulin sensitivity | ↑ respiration |
| Hood et al., 2011 [65] | Overweight, sedentary, non-diabetic | 2 weeks, 3 days/week, HIIT | ↑ HOMA, ↑ glucose transporter protein | ↑ PGC-1α ↑ CS and COX-IV protein |
| Irving et al., 2011 [66] | Non-diabetic offspring of T2D parents and controls, sedentary | 9 days intensive exercise training (continuous moderate and HIIT) | ↑ insulin sensitivity in the controls only | ↑ mitochondrial ATP production ↑ CS activity |
| Hutchison et al., 2012 [67] | Obese insulin-resistant women with PCOS and controls, sedentary | 12 weeks, 3 days/week, moderate intensity and HIIT | ↑ insulin sensitivity | No change in mitochondrial parameters |
| van Tienen et al., 2012 [39] | Obese control, pre-diabetic and T2D | 1 year training in T2D participants (endurance and resistance) | ND | ↑ ATP production ↑ Genes related to TCA cycle, β-oxidation, and oxidative phosphorylation |
| Coen et al., 2015 [68] | Men and women after RYGB surgery | Weight loss only or weight loss and 6 months exercise training (3-5 days/week, moderate intensity) | ↑ glucose tolerance compared to weight loss only group | ↑ respiration in exercise group |
| Konopka et al., 2015 [51] | Obese women with PCOS, and lean insulin-sensitive controls | 12 weeks, 5 days per week, 60 min at 65% VO2 peak | ↑ insulin sensitivity | ↓ H2O2 emission |
| Axelrod et al., 2018 [58] | Obese, pre-diabetic, sedentary, male and female | 12 weeks, 5 days per week, 60 min at 85% HRmax | ↑ insulin sensitivity | ↑ PGC-1α ↑ CS activity |
| Kras et al., 2019 [69] | Obese and non-obese participants, sedentary, male and female | Single exercise session 45 min @ 65% HR reserve | ↑ QUICKI ↓ plasma insulin in non-obese participants | ↑ MAPR in IMF mitochondria, response less in SS mitochondria |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genders, A.J.; Holloway, G.P.; Bishop, D.J. Are Alterations in Skeletal Muscle Mitochondria a Cause or Consequence of Insulin Resistance? Int. J. Mol. Sci. 2020, 21, 6948. https://doi.org/10.3390/ijms21186948
Genders AJ, Holloway GP, Bishop DJ. Are Alterations in Skeletal Muscle Mitochondria a Cause or Consequence of Insulin Resistance? International Journal of Molecular Sciences. 2020; 21(18):6948. https://doi.org/10.3390/ijms21186948
Chicago/Turabian StyleGenders, Amanda J., Graham P. Holloway, and David J. Bishop. 2020. "Are Alterations in Skeletal Muscle Mitochondria a Cause or Consequence of Insulin Resistance?" International Journal of Molecular Sciences 21, no. 18: 6948. https://doi.org/10.3390/ijms21186948
APA StyleGenders, A. J., Holloway, G. P., & Bishop, D. J. (2020). Are Alterations in Skeletal Muscle Mitochondria a Cause or Consequence of Insulin Resistance? International Journal of Molecular Sciences, 21(18), 6948. https://doi.org/10.3390/ijms21186948