T Cell Activation Machinery: Form and Function in Natural and Engineered Immune Receptors


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Key Features of Immune Receptors Involved in T Cell Activation
2.1. The TCR Is a Low-Affinity, High-Sensitivity Immune Receptor
2.2. The TCR Is a Complex of Eight Single-Spanning Membrane Proteins
2.3. Most Lymphocyte-Activating Receptors Have a Similar Multi-Subunit Architecture Based on TM Assembly
2.4. The TCR Contains Signaling Motifs That Are Multiplied Both in Series and in Parallel
2.5. All ITAMs within the TCR Are Not Equal
2.6. Signaling Tails Participate in Complex Interactions with Lipids, Ions and Signaling Partners
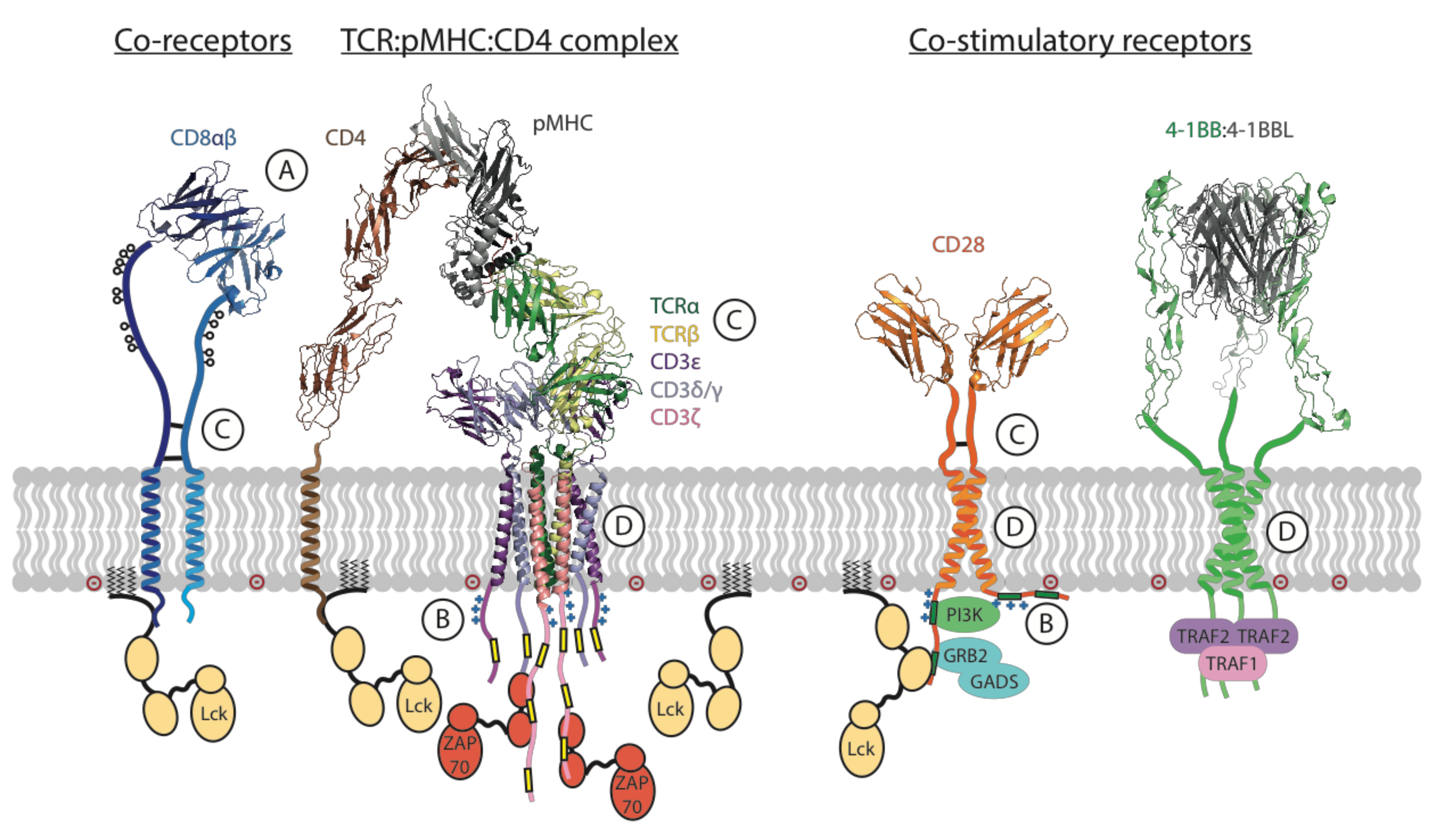
2.7. T Cell Co-Receptors Enhance Sensitivity to Activating Ligands
2.8. Co-Stimulatory and Inhibitory Receptors Control T Cell Priming and Suppression
2.9. Immune Receptor Signaling in the Context of Cell–Cell Interactions
3. Functional Consequences of CAR Design and Structure
3.1. CARs Are High-Affinity, Low-Sensitivity Immune Sensors
3.2. Single-Chain Cars Are Not Monomeric at the Cell Surface
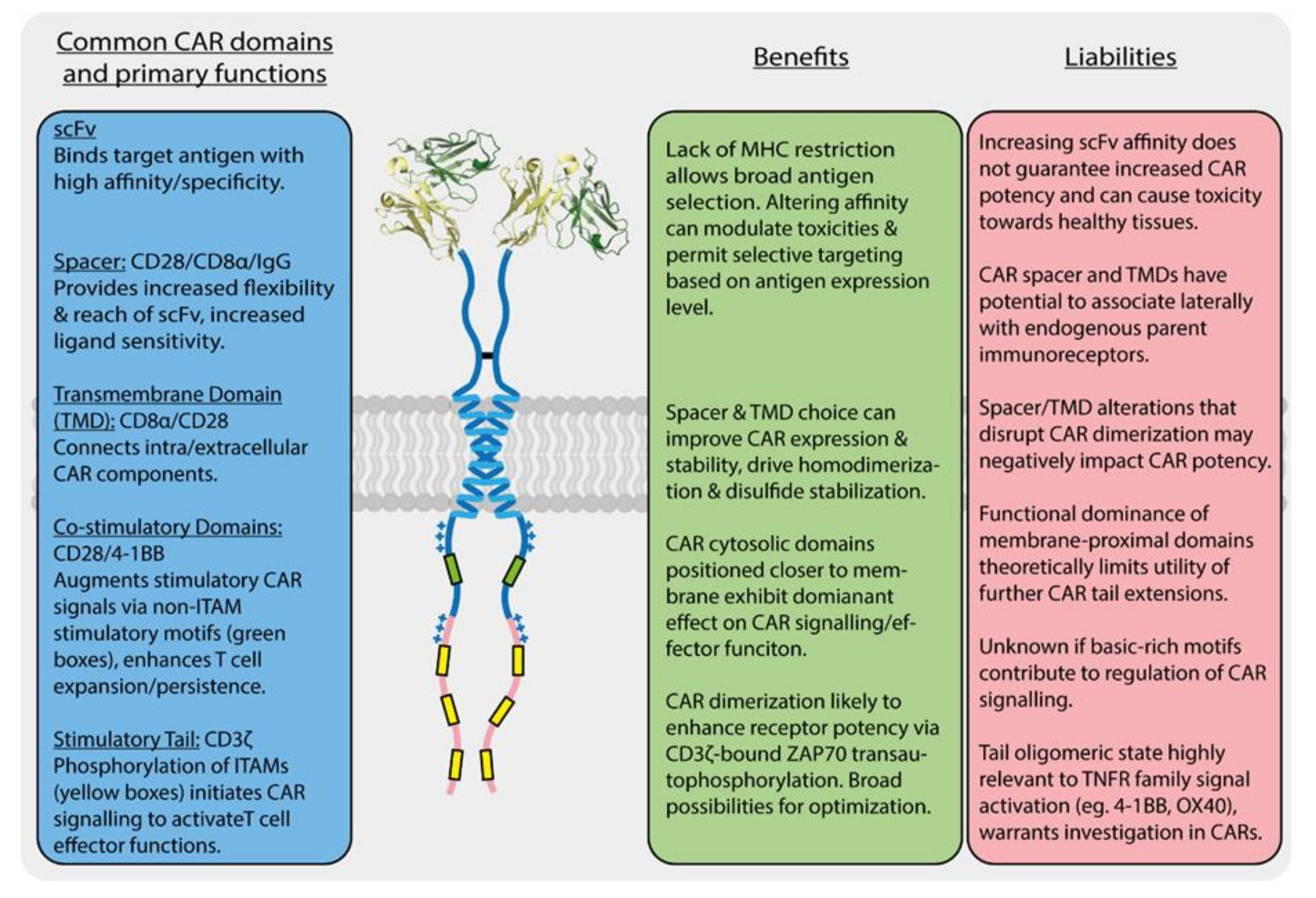
3.3. Repurposing Structural Domains from T Cell Proteins Has Both Benefits and Liabilities
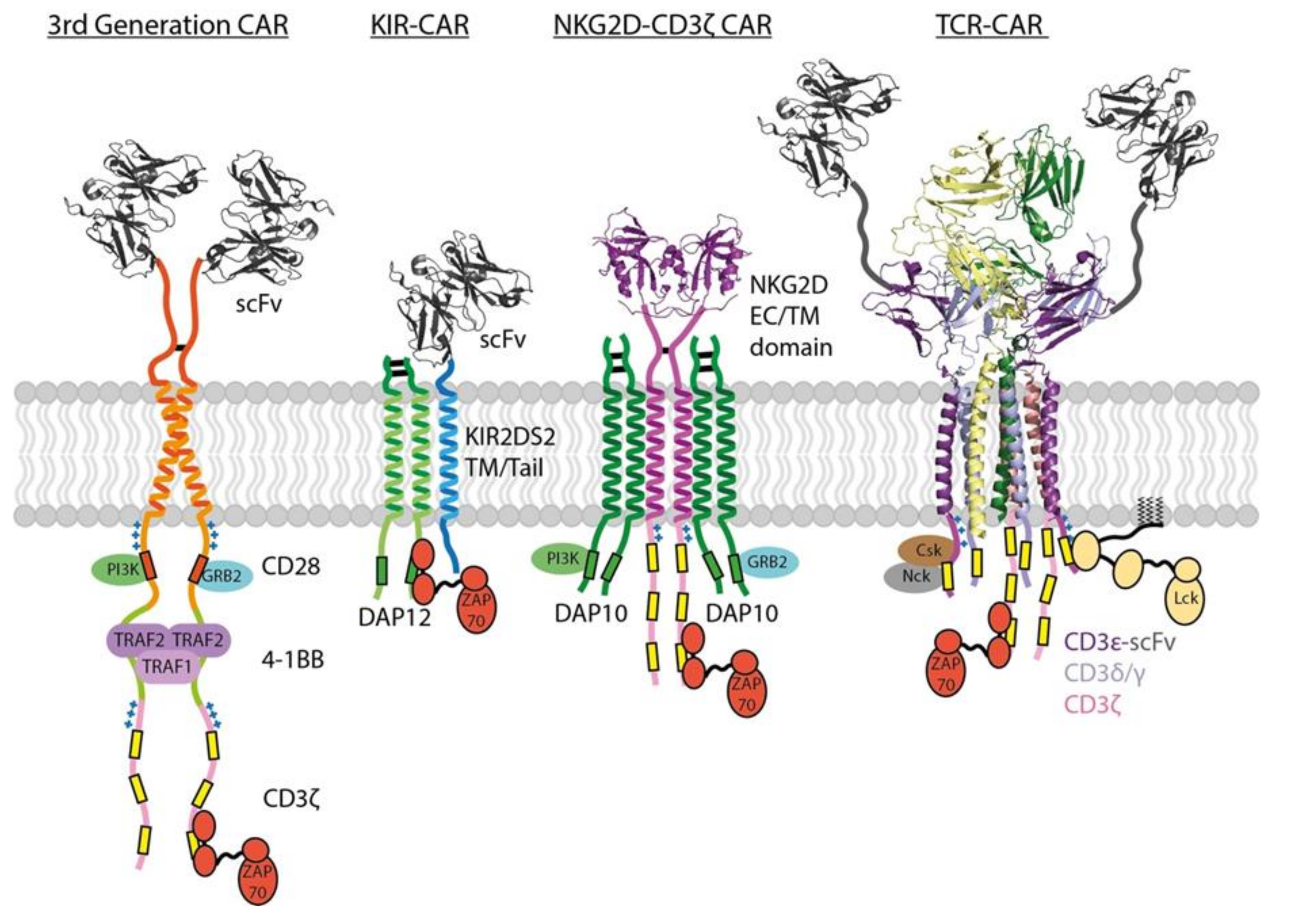
3.4. CAR Potency Is a Function of ITAM Source, Number and Configuration
3.5. CAR Potency Is Also a Function of Co-Stimulatory Motif Source, Number and Configuration
3.6. Additional Modifications to CAR Signalling Tails
3.7. CAR Signalling in the Context of Cell–Cell Interactions
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AICD | Activation-induced Cell death |
| APC | Antigen-presenting cell |
| B-ALL | B-cell Acute Lymphoblastic Leukemia |
| BCR | B-cell receptor |
| BTLA | B- and T-lymphocyte attenuator |
| CAR | Chimeric antigen receptor |
| CRS | Cytokine release syndrome |
| Csk | C-terminal Src kinase |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| Cryo-EM | Cryogenic electron microscopy |
| DAP EC | DNAX-activating protein Extracellular |
| ERK EGFR GADS | Extracellular signal-regulated kinase Epidermal growth factor receptor GRB2-related adaptor protein 2 |
| Grb2 | Growth factor receptor-bound protein 2 |
| HVEM | Herpes virus entry mediator |
| ICOS | Inducible T-cell costimulatory |
| IgSF | Immunoglobulin superfamily |
| IS | Immune Synapse |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| Itk | Interleukin-2-inducible T-cell kinase |
| JAK | Janus kinase |
| KIR | Killer Ig-like receptor |
| KIR2DS2 Lck | Killer Ig-like receptor two Ig domains and short cytoplasmic tail 2 Lymphocyte-specific protein tyrosine kinase |
| MHC | Major histocompatibility complex |
| NK NKG2D | Natural killer Natural killer group 2 member D |
| pMHC | Peptide-bound major histocompatibility complex |
| PDB LFA-1 | Protein Data Bank Lymphocyte function-associated antigen 1 |
| NMR | Nuclear Magnetic Resonance |
| Nck | Non-catalytic region of tyrosine kinase adaptor protein 1 |
| PD-1 PI3K | Programmed death receptor 1 Phosphoinositide 3-kinase |
| PKC | Protein kinase C |
| scFv | Single-chain variable fragment |
| SH2 SHP-1 | Src-homology 2 Src homology 2 domain-containing protein tyrosine phosphatase 1 |
| STAT | Signal Transducer and Activator of Transcription protein |
| TCR | T cell receptor |
| THEMIS TM | Thymocyte-expressed-molecule Transmembrane |
| TNF | Tumor necrosis factor |
| TNFR TNFRSF TRAF | Tumor necrosis factor receptor Tumor necrosis factor receptor Superfamily TNF receptor associated factor |
| ZAP-70 | Zeta-associated protein 70 |
References
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature 2019, 573, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, M.S.; Davis, M.M. TCR Signaling Emerges from the Sum of Many Parts. Front. Immunol. 2012, 3, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yin, Y.; Mariuzza, R.A. Structural and biophysical insights into the role of CD4 and CD8 in T cell activation. Front. Immunol. 2013, 4, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, S.; Azuma, M. The CD28-B7 Family of Co-signaling Molecules. Adv. Exp. Med. Biol. 2019, 1189, 25–51. [Google Scholar] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Clevers, H.; Alarcon, B.; Wileman, T.; Terhorst, C. The T cell receptor/CD3 complex: A dynamic protein ensemble. Annu. Rev. Immunol. 1988, 6, 629–662. [Google Scholar] [CrossRef]
- Klausner, R.D.; Lippincott-Schwartz, J.; Bonifacino, J.S. The T cell antigen receptor: Insights into organelle biology. Annu. Rev. Cell Biol. 1990, 6, 403–431. [Google Scholar] [CrossRef]
- Iwashima, M.; Irving, B.A.; van Oers, N.S.; Chan, A.C.; Weiss, A. Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science 1994, 263, 1136–1139. [Google Scholar] [CrossRef]
- Straus, D.B.; Weiss, A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell 1992, 70, 585–593. [Google Scholar] [CrossRef]
- Rudd, C.; Helms, S.; Barber, E.K.; Schlossman, S.F. The CD4/CD8:p56lck complex in T lymphocytes: A potential mechanism to regulate T-cell growth. Biochem. Cell Biol. 1989, 67, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Trevillyan, J.M.; Dasgupta, J.D.; Wong, L.L.; Schlossman, S.F. The CD4 receptor is complexed in detergent lysates to a protein-tyrosine kinase (pp58) from human T lymphocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 5190–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Annu. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef]
- Goverman, J.; Gomez, S.M.; Segesman, K.D.; Hunkapiller, T.; Laug, W.E.; Hood, L. Chimeric immunoglobulin-T cell receptor proteins form functional receptors: Implications for T cell receptor complex formation and activation. Cell 1990, 60, 929–939. [Google Scholar] [CrossRef]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef]
- Letourneur, F.; Klausner, R.D. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 8905–8909. [Google Scholar] [CrossRef] [Green Version]
- Romeo, C.; Seed, B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell 1991, 64, 1037–1046. [Google Scholar] [CrossRef]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Berry, R.; Call, M.E. Modular Activating Receptors in Innate and Adaptive Immunity. Biochemistry 2017, 56, 1383–1402. [Google Scholar] [CrossRef] [PubMed]
- Love, P.E.; Hayes, S.M. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb. Perspect. Biol. 2010, 2, a002485. [Google Scholar] [CrossRef] [Green Version]
- Mariuzza, R.A.; Agnihotri, P.; Orban, J. The structural basis of T-cell receptor (TCR) activation: An enduring enigma. J. Biol. Chem. 2020, 295, 914–925. [Google Scholar] [CrossRef] [Green Version]
- So, T.; Ishii, N. The TNF-TNFR Family of Co-signal Molecules. Adv. Exp. Med. Biol 2019, 1189, 53–84. [Google Scholar]
- Stone, J.D.; Chervin, A.S.; Kranz, D.M. T-cell receptor binding affinities and kinetics: Impact on T-cell activity and specificity. Immunology 2009, 126, 165–176. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S.; Sansom, D.M. Confusing signals: Recent progress in CTLA-4 biology. Trends Immunol. 2015, 36, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature 1987, 329, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Garboczi, D.N.; Ghosh, P.; Utz, U.; Fan, Q.R.; Biddison, W.E.; Wiley, D.C. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 1996, 384, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.C.; Degano, M.; Stanfield, R.L.; Brunmark, A.; Jackson, M.R.; Peterson, P.A.; Teyton, L.; Wilson, I.A. An alpha beta T cell receptor structure at 2.5 angstrom and its orientation in the TCR-MHC complex. Science 1996, 274, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, R.M.; Doherty, P.C. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 1974, 248, 701–702. [Google Scholar] [CrossRef]
- Irvine, D.J.; Purbhoo, M.A.; Krogsgaard, M.; Davis, M.M. Direct observation of ligand recognition by T cells. Nature 2002, 419, 845–849. [Google Scholar] [CrossRef]
- Huang, J.; Brameshuber, M.; Zeng, X.; Xie, J.; Li, Q.J.; Chien, Y.H.; Valitutti, S.; Davis, M.M. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity 2013, 39, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Purbhoo, M.A.; Irvine, D.J.; Huppa, J.B.; Davis, M.M. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 2004, 5, 524–530. [Google Scholar] [CrossRef]
- Matsui, K.; Boniface, J.J.; Reay, P.A.; Schild, H.; Fazekas de St Groth, B.; Davis, M.M. Low affinity interaction of peptide-MHC complexes with T cell receptors. Science 1991, 254, 1788–1791. [Google Scholar] [CrossRef]
- Weber, S.; Traunecker, A.; Oliveri, F.; Gerhard, W.; Karjalainen, K. Specific low-affinity recognition of major histocompatibility complex plus peptide by soluble T-cell receptor. Nature 1992, 356, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zarnitsyna, V.I.; Liu, B.; Edwards, L.J.; Jiang, N.; Evavold, B.D.; Zhu, C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 2010, 464, 932–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarnitsyna, V.; Zhu, C. T cell triggering: Insights from 2D kinetics analysis of molecular interactions. Phys. Biol. 2012, 9, 045005. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, W.; Evavold, B.D.; Zhu, C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell 2014, 157, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Zhang, T.; Liu, B.; Fei, P.; Cui, L.; Qin, R.; Zhu, H.; Yao, D.; Martinez, R.J.; Hu, W.; et al. Mechano-regulation of Peptide-MHC Class I Conformations Determines TCR Antigen Recognition. Mol. Cell 2019, 73, 1015–1027. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Takeuchi, K.; Sun, Z.Y.; Touma, M.; Castro, C.E.; Fahmy, A.; Lang, M.J.; Wagner, G.; Reinherz, E.L. The alphabeta T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 2009, 284, 31028–31037. [Google Scholar] [CrossRef] [Green Version]
- Reth, M. Antigen receptor tail clue. Nature 1989, 338, 383–384. [Google Scholar] [CrossRef]
- Arnett, K.L.; Harrison, S.C.; Wiley, D.C. Crystal structure of a human CD3-epsilon/delta dimer in complex with a UCHT1 single-chain antibody fragment. Proc. Natl. Acad. Sci. USA 2004, 101, 16268–16273. [Google Scholar] [CrossRef] [Green Version]
- Kjer-Nielsen, L.; Dunstone, M.A.; Kostenko, L.; Ely, L.K.; Beddoe, T.; Mifsud, N.A.; Purcell, A.W.; Brooks, A.G.; McCluskey, J.; Rossjohn, J. Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc. Natl. Acad. Sci. USA 2004, 101, 7675–7680. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.J.; Kim, K.S.; Wagner, G.; Reinherz, E.L. Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 epsilon gamma heterodimer. Cell 2001, 105, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.Y.; Kim, S.T.; Kim, I.C.; Fahmy, A.; Reinherz, E.L.; Wagner, G. Solution structure of the CD3epsilondelta ectodomain and comparison with CD3epsilongamma as a basis for modeling T cell receptor topology and signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 16867–16872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Call, M.E.; Schnell, J.R.; Xu, C.; Lutz, R.A.; Chou, J.J.; Wucherpfennig, K.W. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 2006, 127, 355–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Call, M.E.; Pyrdol, J.; Wiedmann, M.; Wucherpfennig, K.W. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 2002, 111, 967–979. [Google Scholar] [CrossRef] [Green Version]
- Call, M.E.; Wucherpfennig, K.W. The T cell receptor: Critical role of the membrane environment in receptor assembly and function. Annu Rev. Immunol. 2005, 23, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Krshnan, L.; Park, S.; Im, W.; Call, M.J.; Call, M.E. A conserved alphabeta transmembrane interface forms the core of a compact T-cell receptor-CD3 structure within the membrane. Proc. Natl. Acad. Sci. USA 2016, 113, E6649–E6658. [Google Scholar] [CrossRef] [Green Version]
- Kuhns, M.S.; Davis, M.M.; Garcia, K.C. Deconstructing the form and function of the TCR/CD3 complex. Immunity 2006, 24, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Glassman, C.R.; Deshpande, N.R.; Badgandi, H.B.; Parrish, H.L.; Uttamapinant, C.; Stawski, P.S.; Ting, A.Y.; Kuhns, M.S. A Mechanical Switch Couples T Cell Receptor Triggering to the Cytoplasmic Juxtamembrane Regions of CD3zetazeta. Immunity 2015, 43, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Brazin, K.N.; Mallis, R.J.; Das, D.K.; Feng, Y.; Hwang, W.; Wang, J.H.; Wagner, G.; Lang, M.J.; Reinherz, E.L. Structural Features of the alphabetaTCR Mechanotransduction Apparatus That Promote pMHC Discrimination. Front. Immunol. 2015, 6, 441. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Garrity, D.; Call, M.E.; Moffett, H.; Wucherpfennig, K.W. Convergence on a distinctive assembly mechanism by unrelated families of activating immune receptors. Immunity 2005, 22, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. DAP10- and DAP12-associated receptors in innate immunity. Immunol. Rev. 2009, 227, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Call, M.E.; Wucherpfennig, K.W. The assembly of diverse immune receptors is focused on a polar membrane-embedded interaction site. PLoS Biol. 2006, 4, e142. [Google Scholar] [CrossRef] [PubMed]
- Call, M.E.; Wucherpfennig, K.W.; Chou, J.J. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat. Immunol. 2010, 11, 1023–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrity, D.; Call, M.E.; Feng, J.; Wucherpfennig, K.W. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc. Natl. Acad. Sci. USA 2005, 102, 7641–7646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, J.R. Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci. Signal. 2018, 11, eaan1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoGrasso, P.V.; Hawkins, J.; Frank, L.J.; Wisniewski, D.; Marcy, A. Mechanism of activation for Zap-70 catalytic activity. Proc. Natl. Acad. Sci. USA 1996, 93, 12165–12170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.; Wang, H.; Eder, K.D.; Workman, C.J.; Boyd, K.L.; Baquet, Z.; Singh, H.; Forbes, K.; Chruscinski, A.; Smeyne, R.; et al. Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat. Immunol. 2008, 9, 658–666. [Google Scholar] [CrossRef]
- Bettini, M.L.; Chou, P.C.; Guy, C.S.; Lee, T.; Vignali, K.M.; Vignali, D.A.A. Cutting Edge: CD3 ITAM Diversity Is Required for Optimal TCR Signaling and Thymocyte. J. Immunol. 2017, 199, 1555–1560. [Google Scholar] [CrossRef]
- Isakov, N.; Wange, R.L.; Burgess, W.H.; Watts, J.D.; Aebersold, R.; Samelson, L.E. ZAP-70 binding specificity to T cell receptor tyrosine-based activation motifs: The tandem SH2 domains of ZAP-70 bind distinct tyrosine-based activation motifs with varying affinity. J. Exp. Med. 1995, 181, 375–380. [Google Scholar] [CrossRef]
- Osman, N.; Turner, H.; Lucas, S.; Reif, K.; Cantrell, D.A. The protein interactions of the immunoglobulin receptor family tyrosine-based activation motifs present in the T cell receptor zeta subunits and the CD3 gamma, delta and epsilon chains. Eur. J. Immunol. 1996, 26, 1063–1068. [Google Scholar] [CrossRef]
- Zenner, G.; Vorherr, T.; Mustelin, T.; Burn, P. Differential and multiple binding of signal transducing molecules to the ITAMs of the TCR-zeta chain. J. Cell Biochem. 1996, 63, 94–103. [Google Scholar] [CrossRef]
- Xu, C.; Gagnon, E.; Call, M.E.; Schnell, J.R.; Schwieters, C.D.; Carman, C.V.; Chou, J.J.; Wucherpfennig, K.W. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 2008, 135, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aivazian, D.; Stern, L.J. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat. Struct. Biol. 2000, 7, 1023–1026. [Google Scholar] [PubMed]
- Zhang, H.; Cordoba, S.P.; Dushek, O.; van der Merwe, P.A. Basic residues in the T-cell receptor zeta cytoplasmic domain mediate membrane association and modulate signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 19323–19328. [Google Scholar] [PubMed] [Green Version]
- Kuhns, M.S.; Davis, M.M. The safety on the TCR trigger. Cell 2008, 135, 594–596. [Google Scholar]
- Schamel, W.W.; Alarcon, B.; Minguet, S. The TCR is an allosterically regulated macromolecular machinery changing its conformation while working. Immunol. Rev. 2019, 291, 8–25. [Google Scholar]
- Shi, X.; Bi, Y.; Yang, W.; Guo, X.; Jiang, Y.; Wan, C.; Li, L.; Bai, Y.; Guo, J.; Wang, Y.; et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 2013, 493, 111–115. [Google Scholar]
- Li, L.; Guo, X.; Shi, X.; Li, C.; Wu, W.; Yan, C.; Wang, H.; Li, H.; Xu, C. Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E5891–E5899. [Google Scholar]
- Mingueneau, M.; Sansoni, A.; Gregoire, C.; Roncagalli, R.; Aguado, E.; Weiss, A.; Malissen, M.; Malissen, B. The proline-rich sequence of CD3epsilon controls T cell antigen receptor expression on and signaling potency in preselection CD4+CD8+ thymocytes. Nat. Immunol. 2008, 9, 522–532. [Google Scholar]
- Gil, D.; Schamel, W.W.; Montoya, M.; Sanchez-Madrid, F.; Alarcon, B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell 2002, 109, 901–912. [Google Scholar]
- Hartl, F.A.; Beck-Garcia, E.; Woessner, N.M.; Flachsmann, L.J.; Cardenas, R.M.V.; Brandl, S.M.; Taromi, S.; Fiala, G.J.; Morath, A.; Mishra, P.; et al. Noncanonical binding of Lck to CD3epsilon promotes TCR signaling and CAR function. Nat. Immunol. 2020, 21, 902–913. [Google Scholar]
- Wu, W.; Zhou, Q.; Masubuchi, T.; Shi, X.; Li, H.; Xu, X.; Huang, M.; Meng, L.; He, X.; Zhu, H.; et al. Multiple Signaling Roles of CD3epsilon and Its Application in CAR-T Cell Therapy. Cell 2020, 182, 588–871. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.W.; Sun, Z.Y.; Blacklow, S.C.; Wagner, G.; Eck, M.J. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science 2003, 301, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Bijlmakers, M.J.; Isobe-Nakamura, M.; Ruddock, L.J.; Marsh, M. Intrinsic signals in the unique domain target p56(lck) to the plasma membrane independently of CD4. J. Cell Biol. 1997, 137, 1029–1040. [Google Scholar] [CrossRef] [Green Version]
- Casas, J.; Brzostek, J.; Zarnitsyna, V.I.; Hong, J.S.; Wei, Q.; Hoerter, J.A.; Fu, G.; Ampudia, J.; Zamoyska, R.; Zhu, C.; et al. Ligand-engaged TCR is triggered by Lck not associated with CD8 coreceptor. Nat. Commun. 2014, 5, 5624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Huang, J.; Edwards, L.J.; Liu, B.; Zhang, Y.; Beal, C.D.; Evavold, B.D.; Zhu, C. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity 2011, 34, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennecke, S.; Cosson, P. Role of transmembrane domains in assembly and intracellular transport of the CD8 molecule. J. Biol. Chem. 1993, 268, 26607–26612. [Google Scholar]
- Yin, Y.; Wang, X.X.; Mariuzza, R.A. Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc. Natl. Acad. Sci. USA 2012, 109, 5405–5410. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Natarajan, K.; Margulies, D.H. Structural basis of the CD8 alpha beta/MHC class I interaction: Focused recognition orients CD8 beta to a T cell proximal position. J. Immunol. 2009, 183, 2554–2564. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.J.; Ikemizu, S.; Evans, E.J.; Fugger, L.; Bakker, T.R.; van der Merwe, P.A. The nature of molecular recognition by T cells. Nat. Immunol. 2003, 4, 217–224. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Carding, S.; Jones, B.; Murray, J.; Portoles, P.; Rasmussen, R.; Rojo, J.; Saizawa, K.; West, J.; Bottomly, K. CD4+ T cells: Specificity and function. Immunol. Rev. 1988, 101, 39–80. [Google Scholar] [CrossRef]
- Hampl, J.; Chien, Y.H.; Davis, M.M. CD4 augments the response of a T cell to agonist but not to antagonist ligands. Immunity 1997, 7, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Parrish, H.L.; Glassman, C.R.; Keenen, M.M.; Deshpande, N.R.; Bronnimann, M.P.; Kuhns, M.S. A Transmembrane Domain GGxxG Motif in CD4 Contributes to Its Lck-Independent Function but Does Not Mediate CD4 Dimerization. PLoS ONE 2015, 10, e0132333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, M. Co-signal Molecules in T-Cell Activation: Historical Overview and Perspective. Adv. Exp. Med. Biol. 2019, 1189, 3–23. [Google Scholar] [PubMed]
- Weiss, A.; Manger, B.; Imboden, J. Synergy between the T3/antigen receptor complex and Tp44 in the activation of human T cells. J. Immunol. 1986, 137, 819–825. [Google Scholar] [PubMed]
- Porciello, N.; Tuosto, L. CD28 costimulatory signals in T lymphocyte activation: Emerging functions beyond a qualitative and quantitative support to TCR signalling. Cytokine Growth Factor Rev. 2016, 28, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Porciello, N.; Grazioli, P.; Campese, A.F.; Kunkl, M.; Caristi, S.; Mastrogiovanni, M.; Muscolini, M.; Spadaro, F.; Favre, C.; Nunes, J.A.; et al. A non-conserved amino acid variant regulates differential signalling between human and mouse CD28. Nat. Commun. 2018, 9, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Thaker, Y.R.; Raab, M.; Strebhardt, K.; Rudd, C.E. GTPase-activating protein Rasal1 associates with ZAP-70 of the TCR and negatively regulates T-cell tumor immunity. Nat. Commun. 2019, 10, 4804. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef] [Green Version]
- Gaud, G.; Lesourne, R.; Love, P.E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018, 18, 485–497. [Google Scholar] [CrossRef]
- Dobbins, J.; Gagnon, E.; Godec, J.; Pyrdol, J.; Vignali, D.A.A.; Sharpe, A.H.; Wucherpfennig, K.W. Binding of the cytoplasmic domain of CD28 to the plasma membrane inhibits Lck recruitment and signaling. Sci. Signal. 2016, 9, ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Pan, W.; Chen, S.; Trendel, N.; Jiang, S.; Xiao, F.; Xue, M.; Wu, W.; Peng, Z.; Li, X.; et al. Dynamic regulation of CD28 conformation and signaling by charged lipids and ions. Nat. Struct. Mol. Biol. 2017, 24, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lockhart, M.; Kim, M.; Miller, J. Cutting edge: A role for inside-out signaling in TCR regulation of CD28 ligand binding. J. Immunol. 2011, 187, 5515–5519. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lockhart, M.; Rojas, A.V.; Fettis, M.M.; Bauserman, R.; Higa, T.R.; Miao, H.; Waugh, R.E.; Miller, J. T cell receptor signaling can directly enhance the avidity of CD28 ligand binding. PLoS ONE 2014, 9, e89263. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, M.H. Integrin activation. BMB Rep. 2014, 47, 655–659. [Google Scholar] [CrossRef]
- Leddon, S.A.; Fettis, M.M.; Abramo, K.; Kelly, R.; Oleksyn, D.; Miller, J. The CD28 Transmembrane Domain Contains an Essential Dimerization Motif. Front. Immunol. 2020, 11, 1519. [Google Scholar] [CrossRef]
- Blazquez-Moreno, A.; Park, S.; Im, W.; Call, M.J.; Call, M.E.; Reyburn, H.T. Transmembrane features governing Fc receptor CD16A assembly with CD16A signaling adaptor molecules. Proc. Natl. Acad Sci. USA 2017, 114, E5645–e5654. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef]
- Dong, H.D.; Zhu, G.F.; Tamada, K.; Chen, L.P. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.; Chemova, T.; Iwai, Y.; Malenkovich, N.; Long, A.; Bourque, K.; Boussiotis, V.; Nishimura, H.; Honjo, T.; et al. PD-L2, a novel B7 homologue, is a second ligand for PD-1 and inhibits T cell activation. Faseb J. 2001, 15, A345. [Google Scholar]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Lanfranco, A.R.; Braunstein, I.; Riley, J.L. B and T lymphocyte attenuator-mediated signal transduction provides a potent inhibitory signal to primary human CD4 T cells that can be initiated by multiple phosphotyrosine motifs. J. Immunol. 2006, 176, 6603–6614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Z.R.; Shao, X.X.; Ji, X.Y.; Dong, L.H.; Wei, J.C.; Xiong, Z.Q.; Liu, W.L.; Qi, H. Transmembrane domain-mediated Lck association underlies bystander and costimulatory ICOS signaling. Cell Mol. Immunol. 2020, 17, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Yokosuka, T.; Kobayashi, W.; Sakata-Sogawa, K.; Takamatsu, M.; Hashimoto-Tane, A.; Dustin, M.L.; Tokunaga, M.; Saito, T. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity 2008, 29, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Carbone, C.B.; Kern, N.; Fernandes, R.A.; Hui, E.; Su, X.; Garcia, K.C.; Vale, R.D. In vitro reconstitution of T cell receptor-mediated segregation of the CD45 phosphatase. Proc. Natl. Acad. Sci. USA 2017, 114, E9338–E9345. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, K.; Wiseman, D.; Brown, M.H.; Gould, K.; van der Merwe, P.A. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 2005, 436, 578–582. [Google Scholar] [CrossRef]
- Davis, S.J.; van der Merwe, P.A. The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 2006, 7, 803–809. [Google Scholar] [CrossRef]
- James, J.R.; Vale, R.D. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature 2012, 487, 64–69. [Google Scholar] [CrossRef]
- Choudhuri, K.; Parker, M.; Milicic, A.; Cole, D.K.; Shaw, M.K.; Sewell, A.K.; Stewart-Jones, G.; Dong, T.; Gould, K.G.; van der Merwe, P.A. Peptide-major histocompatibility complex dimensions control proximal kinase-phosphatase balance during T cell activation. J. Biol. Chem. 2009, 284, 26096–26105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzer-Attas, C.J.; Schindler, D.G.; Waks, T.; Eshhar, Z. Harnessing Syk family tyrosine kinases as signaling domains for chimeric single chain of the variable domain receptors: Optimal design for T cell activation. J. Immunol. 1998, 160, 145–154. [Google Scholar] [PubMed]
- Moritz, D.; Groner, B. A spacer region between the single chain antibody- and the CD3 zeta-chain domain of chimeric T cell receptor components is required for efficient ligand binding and signaling activity. Gene Ther. 1995, 2, 539–546. [Google Scholar] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintas-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [Green Version]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38. (in press). [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Sievers, N.M.; Dorrie, J.; Schaft, N. CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int. J. Mol. Sci. 2020, 21, 3525. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Drokow, E.K.; Ahmed, H.A.W.; Amponsem-Boateng, C.; Akpabla, G.S.; Song, J.; Shi, M.; Sun, K. Survival outcomes and efficacy of autologous CD19 chimeric antigen receptor-T cell therapy in the patient with diagnosed hematological malignancies: A systematic review and meta-analysis. Ther. Clin. Risk Manag. 2019, 15, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: Interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef]
- Gudipati, V.; Rydzek, J.; Doel-Perez, I.; Goncalves, V.D.R.; Scharf, L.; Konigsberger, S.; Lobner, E.; Kunert, R.; Einsele, H.; Stockinger, H.; et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat. Immunol. 2020, 21, 848–856. [Google Scholar] [CrossRef]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J. Immunol. 2018, 200, 1088–1100. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.J.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; Simonelli, L.; Magnani, C.F.; Biondi, A.; Biagi, E.; Tettamanti, S.; Varani, L. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther. 2017, 25, 1933–1945. [Google Scholar] [CrossRef]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.P.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; de Donk, N.W.C.J.V.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Neelapu, S.S. Managing the toxicities of CAR T-cell therapy. Hematol. Oncol. 2019, 37, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Salzer, B.; Schueller, C.M.; Zajc, C.U.; Peters, T.; Schoeber, M.A.; Kovacic, B.; Buri, M.C.; Lobner, E.; Dushek, O.; Huppa, J.B.; et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun. 2020, 11, 4166. [Google Scholar] [CrossRef]
- Rutledge, T.; Cosson, P.; Manolios, N.; Bonifacino, J.S.; Klausner, R.D. Transmembrane helical interactions: Zeta chain dimerization and functional association with the T cell antigen receptor. Embo J. 1992, 11, 3245–3254. [Google Scholar] [CrossRef]
- Brudno, J.N.; Lam, N.; Vanasse, D.; Shen, Y.W.; Rose, J.J.; Rossi, J.; Xue, A.; Bot, A.; Scholler, N.; Mikkilineni, L.; et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020, 26, 270–280. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [Green Version]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, A.M.; Chui, D.; Reche, P.A.; Priatel, J.J.; Marth, J.D.; Reinherz, E.L. Developmentally regulated glycosylation of the CD8alphabeta coreceptor stalk modulates ligand binding. Cell 2001, 107, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Ramello, M.C.; Benzaid, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabon-Saldana, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, 568. [Google Scholar] [CrossRef]
- Zhang, T.; Lemoi, B.A.; Sentman, C.L. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood 2005, 106, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.; Meehan, K.R.; Sentman, C.L. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor-expressing T cells. Gene Ther. 2011, 18, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, A.; Zhang, T.; DeMars, L.R.; Conejo-Garcia, J.; Roby, K.F.; Sentman, C.L. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007, 67, 5003–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demoulin, B.; Cook, W.J.; Murad, J.; Graber, D.J.; Sentman, M.L.; Lonez, C.; Gilham, D.E.; Sentman, C.L.; Agaugue, S. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Future Oncol. 2017, 13, 1593–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Shin, C.; Kubo, N.; Mihara, K.; Iwabuchi, H.; Takachi, T.; Imamura, M.; Saitoh, A.; Imai, C. Development and characterisation of NKp44-based chimeric antigen receptors that confer T cells with NK cell-like specificity. Clin. Transl. Immunol. 2020, 9, e1147. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wang, L.C.; Tsai, C.Y.; Bhoj, V.; Gershenson, Z.; Moon, E.; Newick, K.; Sun, J.; Lo, A.; Baradet, T.; et al. Generation of Potent T-cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol. Res. 2015, 3, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L.; Corliss, B.C.; Wu, J.; Leong, C.; Phillips, J.H. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 1998, 391, 703–707. [Google Scholar] [CrossRef]
- Haynes, N.M.; Snook, M.B.; Trapani, J.A.; Cerruti, L.; Jane, S.M.; Smyth, M.J.; Darcy, P.K. Redirecting mouse CTL against colon carcinoma: Superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J. Immunol 2001, 166, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Maldini, C.R.; Claiborne, D.T.; Okawa, K.; Chen, T.; Dopkin, D.L.; Shan, X.; Power, K.A.; Trifonova, R.T.; Krupp, K.; Phelps, M.; et al. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat. Med. 2020. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef] [Green Version]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P.; et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Badeti, S.; Dotti, G.; Jiang, J.G.; Wang, H.; Dermody, J.; Soteropoulos, P.; Streck, D.; Birge, R.B.; Liu, C. The Role of Immunological Synapse in Predicting the Efficacy of Chimeric Antigen Receptor (CAR) Immunotherapy. Cell Commun. Signal. 2020, 18, 134. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, R.; Libby, K.A.; Blaeschke, F.; Fuchs, W.; Marson, A.; Vale, R.D.; Su, X. Rewired signaling network in T cells expressing the chimeric antigen receptor (CAR). EMBO J. 2020, 39, e104730. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandler, N.J.; Call, M.J.; Call, M.E. T Cell Activation Machinery: Form and Function in Natural and Engineered Immune Receptors. Int. J. Mol. Sci. 2020, 21, 7424. https://doi.org/10.3390/ijms21197424
Chandler NJ, Call MJ, Call ME. T Cell Activation Machinery: Form and Function in Natural and Engineered Immune Receptors. International Journal of Molecular Sciences. 2020; 21(19):7424. https://doi.org/10.3390/ijms21197424
Chicago/Turabian StyleChandler, Nicholas J., Melissa J. Call, and Matthew E. Call. 2020. "T Cell Activation Machinery: Form and Function in Natural and Engineered Immune Receptors" International Journal of Molecular Sciences 21, no. 19: 7424. https://doi.org/10.3390/ijms21197424
APA StyleChandler, N. J., Call, M. J., & Call, M. E. (2020). T Cell Activation Machinery: Form and Function in Natural and Engineered Immune Receptors. International Journal of Molecular Sciences, 21(19), 7424. https://doi.org/10.3390/ijms21197424