Unconventional Peptide Presentation by Classical MHC Class I and Implications for T and NK Cell Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
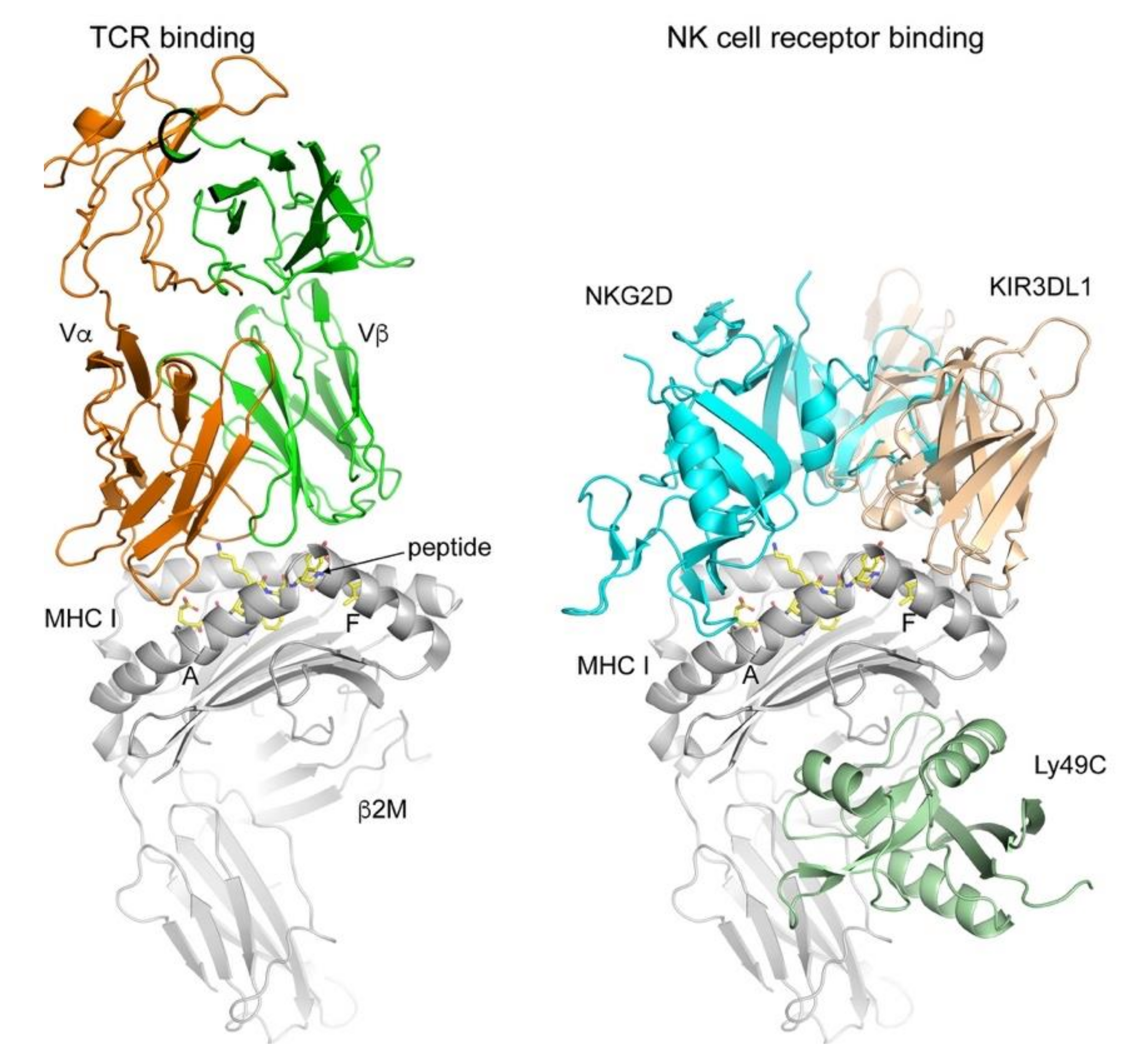
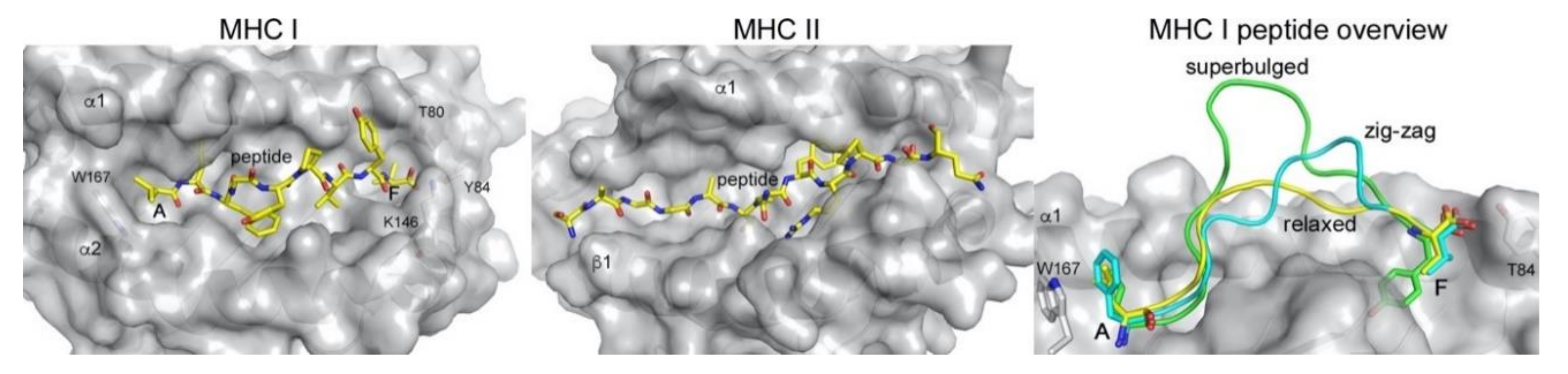
:1. Antigen Presentation by MHC I and II and T Cell Activation
2. NK Cell Recognition of MHC I: A Check and Balance System
3. Unconventional Peptide Presentation
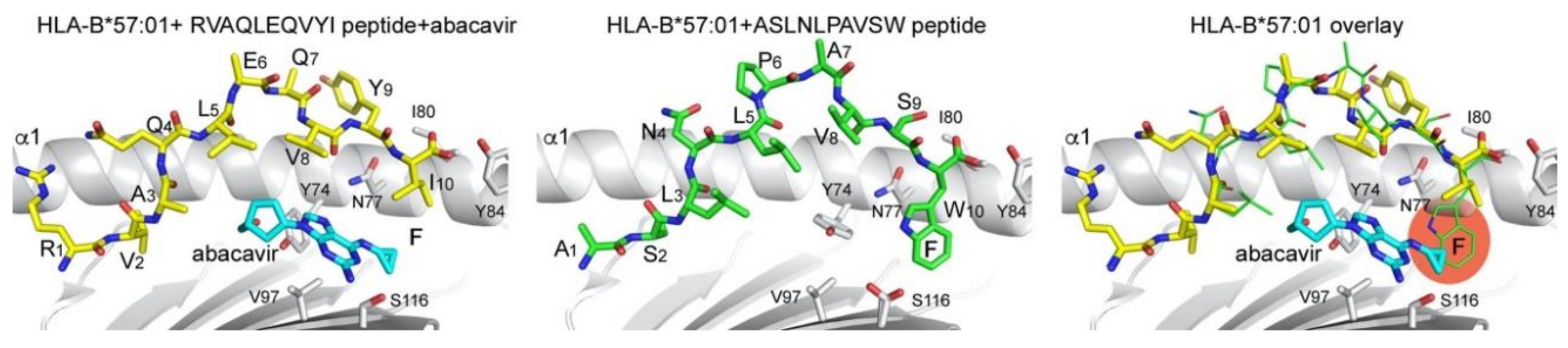
3.1. Small Molecule Modulation of Peptide Presentation
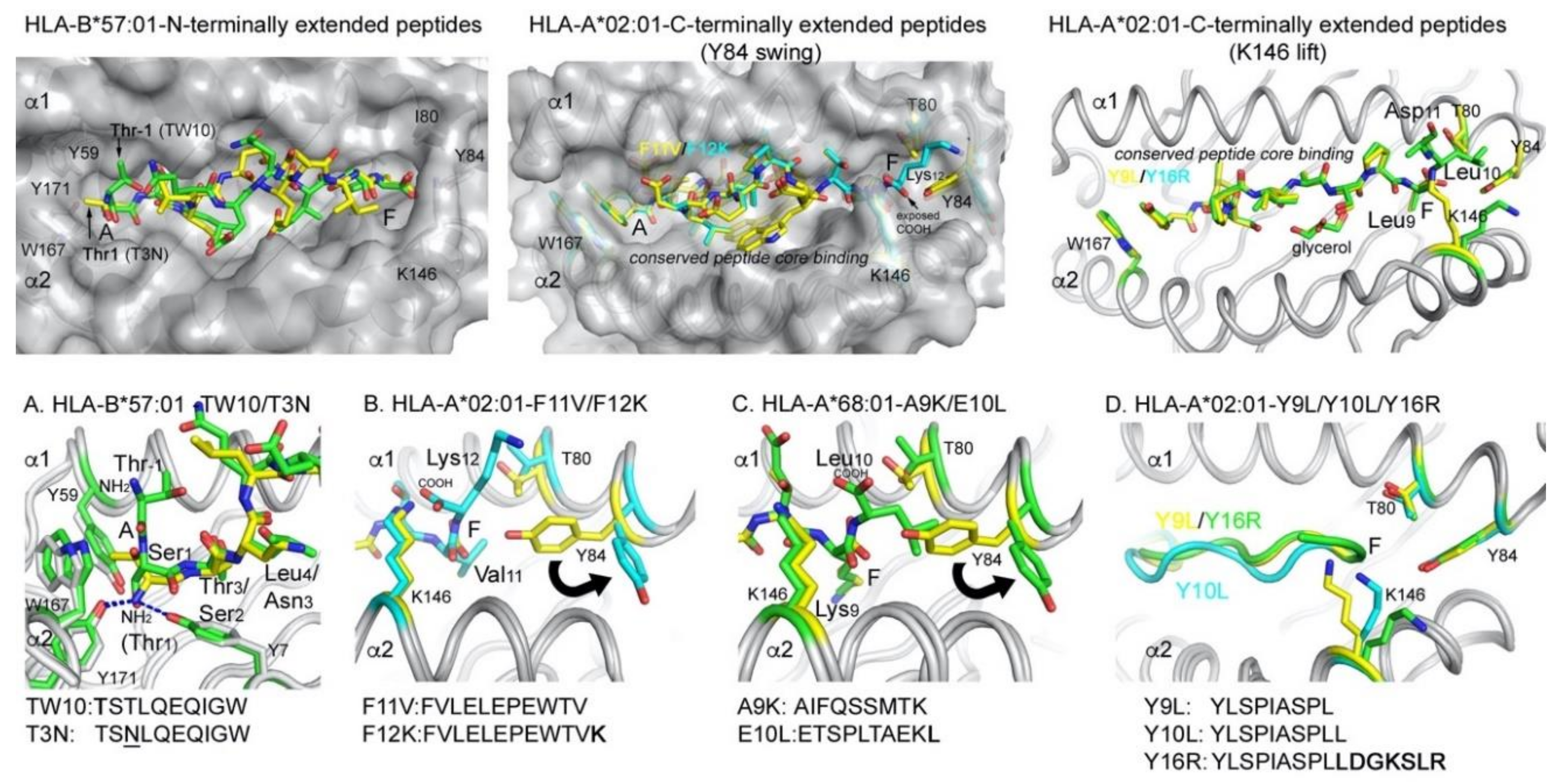
3.2. Unconventional Presentation of N- and C-Terminally Extended Peptides
3.2.1. Presentation of C-Terminally Extending Peptides
3.2.2. Presentation of N-Terminally Extending Peptides
4. Killer Immunoglobulin Receptors (KIRs) in Infection and Cancer
5. Outlook and Future Perspectives
6. Patents
Funding
Conflicts of Interest
Abbreviations
| MHC | Major Histocompatibility |
| HLA | Human Leukocyte Antigen |
| KIR | Killer Immunoglobulin Receptor |
| APC | Antigen presenting cell |
| pMHC | Peptide MHC |
References
- Adams, E.J.; Luoma, A.M. The adaptable major histocompatibility complex (MHC) fold: Structure and function of nonclassical and MHC class I-like molecules. Annu. Rev. Immunol. 2013, 31, 529–561. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Gravano, D.M.; Hoyer, K.K. Promotion and prevention of autoimmune disease by CD8+ T cells. J. Autoimmun. 2013, 45, 68–79. [Google Scholar] [CrossRef]
- Kumar, V.; Sercarz, E. Induction or protection from experimental autoimmune encephalomyelitis depends on the cytokine secretion profile of TCR peptide-specific regulatory CD4 T cells. J. Immunol. 1998, 161, 6585–6591. [Google Scholar]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Grotzke, J.E.; Sengupta, D.; Lu, Q.; Cresswell, P. The ongoing saga of the mechanism(s) of MHC class I-restricted cross-presentation. Curr. Opin. Immunol. 2017, 46, 89–96. [Google Scholar] [CrossRef]
- Munz, C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 2016, 272, 17–27. [Google Scholar] [CrossRef]
- Unanue, E.R.; Turk, V.; Neefjes, J. Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annu. Rev. Immunol. 2016, 34, 265–297. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 2001, 1, 126–134. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 2001, 19, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Huppa, J.B.; Davis, M.M. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 2003, 3, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Pettmann, J.; Santos, A.M.; Dushek, O.; Davis, S.J. Membrane Ultrastructure and T Cell Activation. Front. Immunol. 2018, 9, 2152. [Google Scholar] [CrossRef] [PubMed]
- Verboogen, D.R.; Dingjan, I.; Revelo, N.H.; Visser, L.J.; ter Beest, M.; van den Bogaart, G. The dendritic cell side of the immunological synapse. Biomol. Concepts 2016, 7, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromley, S.K.; Burack, W.R.; Johnson, K.G.; Somersalo, K.; Sims, T.N.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse. Annu. Rev. Immunol. 2001, 19, 375–396. [Google Scholar] [CrossRef]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef]
- Malissen, B.; Bongrand, P. Early T cell activation: Integrating biochemical, structural, and biophysical cues. Annu. Rev. Immunol. 2015, 33, 539–561. [Google Scholar] [CrossRef]
- Garcia, K.C.; Degano, M.; Stanfield, R.L.; Brunmark, A.; Jackson, M.R.; Peterson, P.A.; Teyton, L.; Wilson, I.A. An alphabeta T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science 1996, 274, 209–219. [Google Scholar] [CrossRef]
- Li, P.; Morris, D.L.; Willcox, B.E.; Steinle, A.; Spies, T.; Strong, R.K. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat. Immunol. 2001, 2, 443–451. [Google Scholar] [CrossRef]
- Sullivan, L.C.; Berry, R.; Sosnin, N.; Widjaja, J.M.; Deuss, F.A.; Balaji, G.R.; LaGruta, N.L.; Mirams, M.; Trapani, J.A.; Rossjohn, J.; et al. Recognition of the Major Histocompatibility Complex (MHC) Class Ib Molecule H2-Q10 by the Natural Killer Cell Receptor Ly49C. J. Biol. Chem. 2016, 291, 18740–18752. [Google Scholar] [CrossRef] [Green Version]
- Vivian, J.P.; Duncan, R.C.; Berry, R.; O’Connor, G.M.; Reid, H.H.; Beddoe, T.; Gras, S.; Saunders, P.M.; Olshina, M.A.; Widjaja, J.M.L.; et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature 2011, 479, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Madden, D.R. The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol. 1995, 13, 587–622. [Google Scholar] [CrossRef] [PubMed]
- Fremont, D.H.; Matsumura, M.; Stura, E.A.; Peterson, P.A.; Wilson, I.A. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb. Science 1992, 257, 919–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.C.; Jardetzky, T.S.; Garrett, T.P.; Lane, W.S.; Strominger, J.L.; Wiley, D.C. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature 1992, 360, 364–366. [Google Scholar] [CrossRef]
- Zhang, W.; Young, A.C.; Imarai, M.; Nathenson, S.G.; Sacchettini, J.C. Crystal structure of the major histocompatibility complex class I H-2Kb molecule containing a single viral peptide: Implications for peptide binding and T-cell receptor recognition. Proc. Natl. Acad. Sci. USA 1992, 89, 8403–8407. [Google Scholar] [CrossRef] [Green Version]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Roder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2007, 2, e796. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Dow, C.; Mothe, B.; Sette, A.; Peters, B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008, 4, e1000048. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brightman, S.E.; Naradikian, M.S.; Miller, A.M.; Schoenberger, S.P. Harnessing neoantigen specific CD4 T cells for cancer immunotherapy. J. Leukoc. Biol. 2020, 107, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Forget, M.A.; Haymaker, C.; Hess, K.R.; Meng, Y.J.; Creasy, C.; Karpinets, T.; Fulbright, O.J.; Roszik, J.; Woodman, S.E.; Kim, Y.U.; et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin. Cancer Res. 2018, 24, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; Hendel, H.W.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: A pilot study. Oncoimmunology 2018, 7, e1502905. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Petrie, E.J.; Clements, C.S.; Lin, J.; Sullivan, L.C.; Johnson, D.; Huyton, T.; Heroux, A.; Hoare, H.L.; Beddoe, T.; Reid, H.H.; et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J. Exp. Med. 2008, 205, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Verweij, M.C.; Horst, D.; Griffin, B.D.; Luteijn, R.D.; Davison, A.J.; Ressing, M.E.; Wiertz, E.J. Viral inhibition of the transporter associated with antigen processing (TAP): A striking example of functional convergent evolution. PLoS Pathog. 2015, 11, e1004743. [Google Scholar] [CrossRef] [Green Version]
- Piguet, V. Receptor modulation in viral replication: HIV, HSV, HHV-8 and HPV: Same goal, different techniques to interfere with MHC-I antigen presentation. Curr. Top. Microbiol. Immunol. 2005, 285, 199–217. [Google Scholar]
- Hegde, N.R.; Chevalier, M.S.; Johnson, D.C. Viral inhibition of MHC class II antigen presentation. Trends Immunol. 2003, 24, 278–285. [Google Scholar] [CrossRef]
- Fruh, K.; Ahn, K.; Peterson, P.A. Inhibition of MHC class I antigen presentation by viral proteins. J. Mol. Med. 1997, 75, 18–27. [Google Scholar] [CrossRef]
- Sutherland, C.L.; Chalupny, N.J.; Cosman, D. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 2001, 181, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Borrego, F.; Ulbrecht, M.; Weiss, E.H.; Coligan, J.E.; Brooks, A.G. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J. Exp. Med. 1998, 187, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Pizarro, J.C.; Kerns, J.; Strong, R.K. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc. Natl. Acad. Sci. USA 2008, 105, 6696–6701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, H.; Matsumoto, N.; Maenaka, K.; Suzuki, K.; Yamamoto, K. The inhibitory NK cell receptor CD94/NKG2A and the activating receptor CD94/NKG2C bind the top of HLA-E through mostly shared but partly distinct sets of HLA-E residues. Eur. J. Immunol. 2004, 34, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.H.; Faulds, D. Abacavir. Drugs 1998, 55, 729–736, discussion 737–728. [Google Scholar] [CrossRef] [PubMed]
- Hetherington, S.; Hughes, A.R.; Mosteller, M.; Shortino, D.; Baker, K.L.; Spreen, W.; Lai, E.; Davies, K.; Handley, A.; Dow, D.J.; et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 2002, 359, 1121–1122. [Google Scholar] [CrossRef]
- Mallal, S.; Nolan, D.; Witt, C.; Masel, G.; Martin, A.M.; Moore, C.; Sayer, D.; Castley, A.; Mamotte, C.; Maxwell, D.; et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002, 359, 727–732. [Google Scholar] [CrossRef]
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012, 486, 554–558. [Google Scholar] [CrossRef]
- Ostrov, D.A.; Grant, B.J.; Pompeu, Y.A.; Sidney, J.; Harndahl, M.; Southwood, S.; Oseroff, C.; Lu, S.; Jakoncic, J.; de Oliveira, C.A.; et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. USA 2012, 109, 9959–9964. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.H.; Hung, S.I.; Hong, H.S.; Hsih, M.S.; Yang, L.C.; Ho, H.C.; Wu, J.Y.; Chen, Y.T. Medical genetics: A marker for Stevens-Johnson syndrome. Nature 2004, 428, 486. [Google Scholar] [CrossRef] [PubMed]
- McCormack, M.; Alfirevic, A.; Bourgeois, S.; Farrell, J.J.; Kasperaviciute, D.; Carrington, M.; Sills, G.J.; Marson, T.; Jia, X.; de Bakker, P.I.; et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 2011, 364, 1134–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simper, G.S.; Graser, L.S.; Celik, A.A.; Kuhn, J.; Kunze-Schumacher, H.; Ho, G.T.; Blasczyk, R.; Pich, A.; Bade-Doeding, C. The Mechanistic Differences in HLA-Associated Carbamazepine Hypersensitivity. Pharmaceutics 2019, 11, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, T.M.; Chung, W.H.; Wei, C.Y.; Shih, H.Y.; Chen, J.K.; Lin, C.H.; Chen, Y.T.; Hung, S.I. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. J. Allergy Clin. Immunol. 2011, 128, 1266–1276.e11. [Google Scholar] [CrossRef] [PubMed]
- Simper, G.S.; Ho, G.T.; Celik, A.A.; Huyton, T.; Kuhn, J.; Kunze-Schumacher, H.; Blasczyk, R.; Bade-Doding, C. Carbamazepine-Mediated Adverse Drug Reactions: CBZ-10,11-epoxide but Not Carbamazepine Induces the Alteration of Peptides Presented by HLA-B *15:02. J. Immunol. Res. 2018, 2018, 5086503. [Google Scholar] [CrossRef] [Green Version]
- Collins, E.J.; Garboczi, D.N.; Wiley, D.C. Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature 1994, 371, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.R.; Castano, A.R.; Peterson, P.A.; Slaughter, C.; Lindahl, K.F.; Deisenhofer, J. Nonclassical binding of formylated peptide in crystal structure of the MHC class Ib molecule H2-M3. Cell 1995, 82, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Horig, H.; Young, A.C.; Papadopoulos, N.J.; DiLorenzo, T.P.; Nathenson, S.G. Binding of longer peptides to the H-2Kb heterodimer is restricted to peptides extended at their C terminus: Refinement of the inherent MHC class I peptide binding criteria. J. Immunol. 1999, 163, 4434–4441. [Google Scholar]
- Motozono, C.; Pearson, J.A.; De Leenheer, E.; Rizkallah, P.J.; Beck, K.; Trimby, A.; Sewell, A.K.; Wong, F.S.; Cole, D.K. Distortion of the Major Histocompatibility Complex Class I Binding Groove to Accommodate an Insulin-derived 10-Mer Peptide. J. Biol. Chem. 2015, 290, 18924–18933. [Google Scholar] [CrossRef] [Green Version]
- McMurtrey, C.; Trolle, T.; Sansom, T.; Remesh, S.G.; Kaever, T.; Bardet, W.; Jackson, K.; McLeod, R.; Sette, A.; Nielsen, M.; et al. Toxoplasma gondii peptide ligands open the gate of the HLA class I binding groove. eLife 2016, 5. [Google Scholar] [CrossRef]
- Remesh, S.G.; Andreatta, M.; Ying, G.; Kaever, T.; Nielsen, M.; McMurtrey, C.; Hildebrand, W.; Peters, B.; Zajonc, D.M. Unconventional Peptide Presentation by Major Histocompatibility Complex (MHC) Class I Allele HLA-A*02:01: BREAKING CONFINEMENT. J. Biol. Chem. 2017, 292, 5262–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaume, P.; Picaud, S.; Baumgaertner, P.; Montandon, N.; Schmidt, J.; Speiser, D.E.; Coukos, G.; Bassani-Sternberg, M.; Filippakopoulos, P.; Gfeller, D. The C-terminal extension landscape of naturally presented HLA-I ligands. Proc. Natl. Acad. Sci. USA 2018, 115, 5083–5088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pymm, P.; Illing, P.T.; Ramarathinam, S.H.; O’Connor, G.M.; Hughes, V.A.; Hitchen, C.; Price, D.A.; Ho, B.K.; McVicar, D.W.; Brooks, A.G.; et al. MHC-I peptides get out of the groove and enable a novel mechanism of HIV-1 escape. Nat. Struct. Mol. Biol. 2017, 24, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulberger, C.L.; McMurtrey, C.P.; Holzemer, A.; Neu, K.E.; Liu, V.; Steinbach, A.M.; Garcia-Beltran, W.F.; Sulak, M.; Jabri, B.; Lynch, V.J.; et al. Human Leukocyte Antigen F Presents Peptides and Regulates Immunity through Interactions with NK Cell Receptors. Immunity 2017, 46, 1018–1029.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Anfossi, N. Inhibitory NK-cell receptors on T cells: Witness of the past, actors of the future. Nat. Rev. Immunol. 2004, 4, 190–198. [Google Scholar] [CrossRef]
- Parham, P. MHC class I molecules and KIRs in human history, health and survival. Nat. Rev. Immunol. 2005, 5, 201–214. [Google Scholar] [CrossRef]
- Thielens, A.; Vivier, E.; Romagné, F. NK cell MHC class I specific receptors (KIR): From biology to clinical intervention. Curr. Opin. Immunol. 2012, 24, 239–245. [Google Scholar] [CrossRef]
- Boyington, J.C.; Motyka, S.A.; Schuck, P.; Brooks, A.G.; Sun, P.D. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature 2000, 405, 537–543. [Google Scholar] [CrossRef]
- Fan, Q.R.; Long, E.O.; Wiley, D.C. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat. Immunol. 2001, 2, 452–460. [Google Scholar] [CrossRef]
- Alter, G.; Martin, M.P.; Teigen, N.; Carr, W.H.; Suscovich, T.J.; Schneidewind, A.; Streeck, H.; Waring, M.; Meier, A.; Brander, C.; et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J. Exp. Med. 2007, 204, 3027–3036. [Google Scholar] [CrossRef] [Green Version]
- Alter, G.; Rihn, S.; Walter, K.; Nolting, A.; Martin, M.; Rosenberg, E.S.; Miller, J.S.; Carrington, M.; Altfeld, M. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J. Virol. 2009, 83, 6798–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrington, M.; Martin, M.P.; van Bergen, J. KIR-HLA intercourse in HIV disease. Trends Microbiol. 2008, 16, 620–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakoo, S.I.; Thio, C.L.; Martin, M.P.; Brooks, C.R.; Gao, X.; Astemborski, J.; Cheng, J.; Goedert, J.J.; Vlahov, D.; Hilgartner, M.; et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 2004, 305, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Castineira, J.R.; Lopez-Vazquez, A.; Diaz-Pena, R.; Alonso-Arias, R.; Martinez-Borra, J.; Perez, R.; Fernandez-Suarez, J.; Melon, S.; Prieto, J.; Rodrigo, L.; et al. Effect of killer immunoglobulin-like receptors in the response to combined treatment in patients with chronic hepatitis C virus infection. J. Virol. 2010, 84, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlenstiel, G.; Martin, M.P.; Gao, X.; Carrington, M.; Rehermann, B. Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J. Clin. Investig. 2008, 118, 1017–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moesta, A.K.; Norman, P.J.; Yawata, M.; Yawata, N.; Gleimer, M.; Parham, P. Synergistic polymorphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J. Immunol. 2008, 180, 3969–3979. [Google Scholar] [CrossRef] [PubMed]
- Terme, M.; Ullrich, E.; Delahaye, N.F.; Chaput, N.; Zitvogel, L. Natural killer cell-directed therapies: Moving from unexpected results to successful strategies. Nat. Immunol. 2008, 9, 486–494. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Chang, C.C.; Campoli, M.; Ferrone, S. Classical and nonclassical HLA class I antigen and NK Cell-activating ligand changes in malignant cells: Current challenges and future directions. Adv. Cancer Res. 2005, 93, 189–234. [Google Scholar] [CrossRef]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Johansson, S.E.; Hejdeman, B.; Hinkula, J.; Johansson, M.H.; Romagné, F.; Wahren, B.; Wagtmann, N.R.; Kärre, K.; Berg, L. NK cell activation by KIR-binding antibody 1-7F9 and response to HIV-infected autologous cells in viremic and controller HIV-infected patients. Clin. Immunol. 2010, 134, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M.; Bakan, C.E.; Zhang, S.; Collins, S.M.; Liang, J.; Srivastava, S.; Hofmeister, C.C.; Efebera, Y.; André, P.; Romagné, F.; et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood 2011, 118, 6387–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.M.; Krogsgaard, M.; Huse, M.; Huppa, J.; Lillemeier, B.F.; Li, Q.J. T cells as a self-referential, sensory organ. Annu. Rev. Immunol. 2007, 25, 681–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zajonc, D.M. Unconventional Peptide Presentation by Classical MHC Class I and Implications for T and NK Cell Activation. Int. J. Mol. Sci. 2020, 21, 7561. https://doi.org/10.3390/ijms21207561
Zajonc DM. Unconventional Peptide Presentation by Classical MHC Class I and Implications for T and NK Cell Activation. International Journal of Molecular Sciences. 2020; 21(20):7561. https://doi.org/10.3390/ijms21207561
Chicago/Turabian StyleZajonc, Dirk M. 2020. "Unconventional Peptide Presentation by Classical MHC Class I and Implications for T and NK Cell Activation" International Journal of Molecular Sciences 21, no. 20: 7561. https://doi.org/10.3390/ijms21207561
APA StyleZajonc, D. M. (2020). Unconventional Peptide Presentation by Classical MHC Class I and Implications for T and NK Cell Activation. International Journal of Molecular Sciences, 21(20), 7561. https://doi.org/10.3390/ijms21207561