1. Introduction
The liver is an organ that spans a large variety of different functions in the body: energy metabolism, detoxification, and production of serum proteins and bile, just to mention a few [
1]. Consequently, liver disease or liver toxicity that cause impaired liver functionality have severe effects on normal body functions. Liver diseases are a major burden to the public health and estimations show a global liver-related mortality rate of approx. 2 million patients per year [
2]. In addition, the liver is one of the organs with the highest susceptibility to drug toxicity, significantly contributing to the high attrition rates in current drug discovery processes [
3]. Therefore, there is a strong need for better and more predictive in vitro models for liver disease and toxicity. A general opinion within the field of safety assessment and disease modeling is that in vitro cell models will increasingly contribute to improve the mechanistic understanding of diseases and the prediction of adverse effects of drugs in humans in the future.
The gold standard for studying liver disease or hepatotoxicity in vitro are metabolically competent human primary hepatocytes (hphep), either freshly isolated or cryopreserved. However, the hphep have several shortcomings that limit their use. The shortage of relevant human donor material, the large donor variation, and the rapid loss of their functionality, e.g., of the expression of the drug metabolizing machinery, in culture [
4,
5,
6,
7], are the most prominent problems. Furthermore, the liver consists of multiple cell types; hepatocytes and non-parenchymal cells, such as cholangiocytes, endothelial cells, hepatic stellate cells, and Kupffer cells. As a consequence, an in vitro liver model with capacity to display all hepatic functions needs to consist of several if not all hepatic cell types and not only hepatocytes. In addition to using multiple cell types, a 3-dimensional (3D) culture environment is beneficial for an in vitro model as it allows the formation of cell-cell contacts and the formation of cell polarity in hepatocytes as indicated by the presence of bile canaliculi [
8].
Nonetheless, functional hepatocytes will constitute the major part of a relevant in vitro liver model, and therefore, stem cell research has been focused on deriving mature hepatocytes from stem cells. Due to the many diverse features of hepatocytes, and also considering the fact that primary hepatocytes rapidly lose key functions when cultured in conventional 2D cultures in vitro, it is not surprising that hepatocytes are one of the most challenging cell type to derive from stem cells and further mature in vitro.
In order to explore the potential of a cell model and identify good-to-go areas for the cell model, a thorough characterization of the cells in question is necessary. This characterization needs to be done on multiple levels, preferably combining large-scale transcriptomics assessment with multiple functional assays [
9]. Importantly, hepatocytes exist in periportal and perivenous phenotypes in the liver lobe, commonly known as metabolic zonation [
10], and it is important to look at features for both these hepatocyte populations when assessing a hepatocyte cell model.
Human induced pluripotent stem cells (hiPSC) are a virtually unlimited source of cells and have the potential to differentiate into specialized cell types, which provides unique opportunities for usage in a wide range of applications. The possibility to convert hiPSCs into functional hepatocytes allows for novel opportunities in assay development and holds great potential for future pre-clinical and regenerative medicine applications. For example, hiPSC derived from patients suffering from, e.g., inherited metabolic diseases can be used for modeling these types of diseases in vitro [
11,
12,
13]. Combining the hiPSC technology with the constantly progressing genome editing technologies, e.g., CRISPR-Cas9, further increases the potential for disease modeling and therapeutic applications [
14,
15].
The study presented here describes a thorough characterization of hiPSC-derived hepatocytes (hiPS-HEP), in order to assess their potential as in vitro tools for metabolism studies and disease modeling. The cells were subjected to a broad panel of characterization assays, ranging from transcriptomics analysis, protein expression, to multiple functional assays. The results show that the hiPS-HEP possess many adult hepatocyte features that can be maintained in conventional 2D cultures for over 2 weeks. Furthermore, primary hepatic stellate cells could be activated in 2D co-cultures with hiPS-HEP which, to the best of our knowledge, is the first time that this is reported. In addition, 3D co-culture spheroids were established, paving the way for using these for modeling non-alcoholic steatohepatitis (NASH). Importantly, the transcriptomics analysis revealed that a majority of genes (86%) are similarly expressed in all the three test groups. Taken together, this study highlights the potential of hiPSC-derived hepatocytes as a robust and reproducible source for hepatocyte-related in vitro models, e.g., metabolism-related liver diseases.
3. Discussion
The present study covers an in-depth characterization of hiPSC-derived hepatocytes including multiple phenotypic and functional aspects of relevance for several application areas, for example modeling of metabolic disease.
An essential requirement for the utility of stem cell-derived hepatocytes for all application areas is that cultures with a very high hepatocyte content can be robustly derived from large panels of stem cell lines. To evaluate the hepatocyte content in the hiPS-HEP cultures, we stained for the key hepatocyte transcription factor HNF4α and found that it was expressed in 92% of cells in the hiPS-HEP cultures, thus indicating a highly efficient differentiation result and a near-homogeneous culture regarding HNF4α expression. A similarly high differentiation efficiency, 93.8% HNF4α-positive cells, was previously shown when we screened a panel of 25 human pluripotent stem cell lines using an earlier version of the hepatocyte differentiation protocol used in this study to derive hiPS-HEP [
16]. Notably, the hepatocyte differentiation protocol worked for all lines without any modifications. In contrast to our findings, others have reported that their protocols either do not work for some hPSC lines making it necessary to select lines that appear more prone to differentiate into hepatocytes [
23], or that they require time- and labor-intensive adaptations of the protocol for individual lines [
24]. One great advantage of the hiPSC technology is the possibility to generate iPSC lines from many different donors, healthy and diseased, so that one can recreate the population diversity in vitro. However, this requires differentiation protocols that, without adaptations, generate highly pure hepatocyte cultures from virtually all lines.
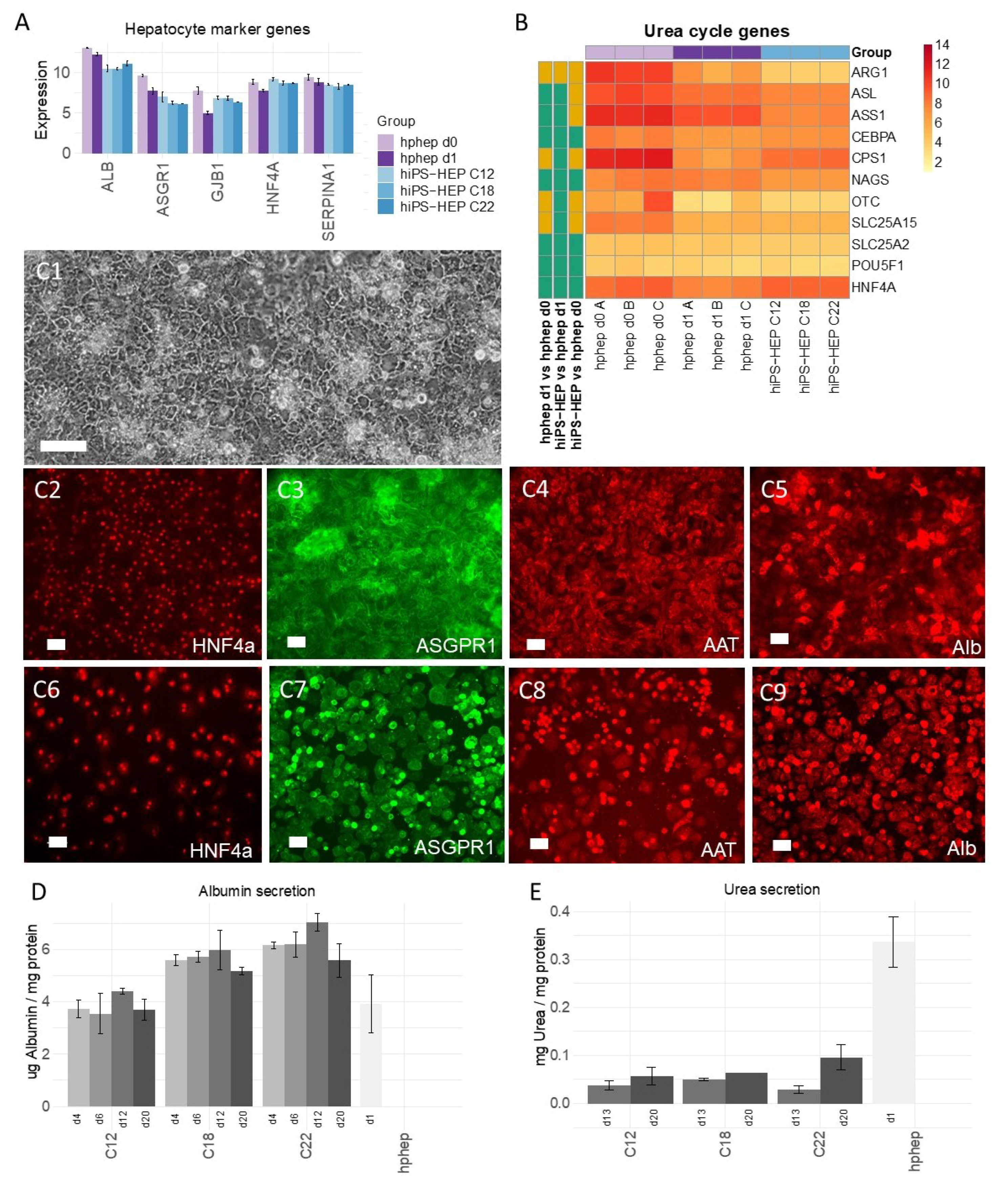
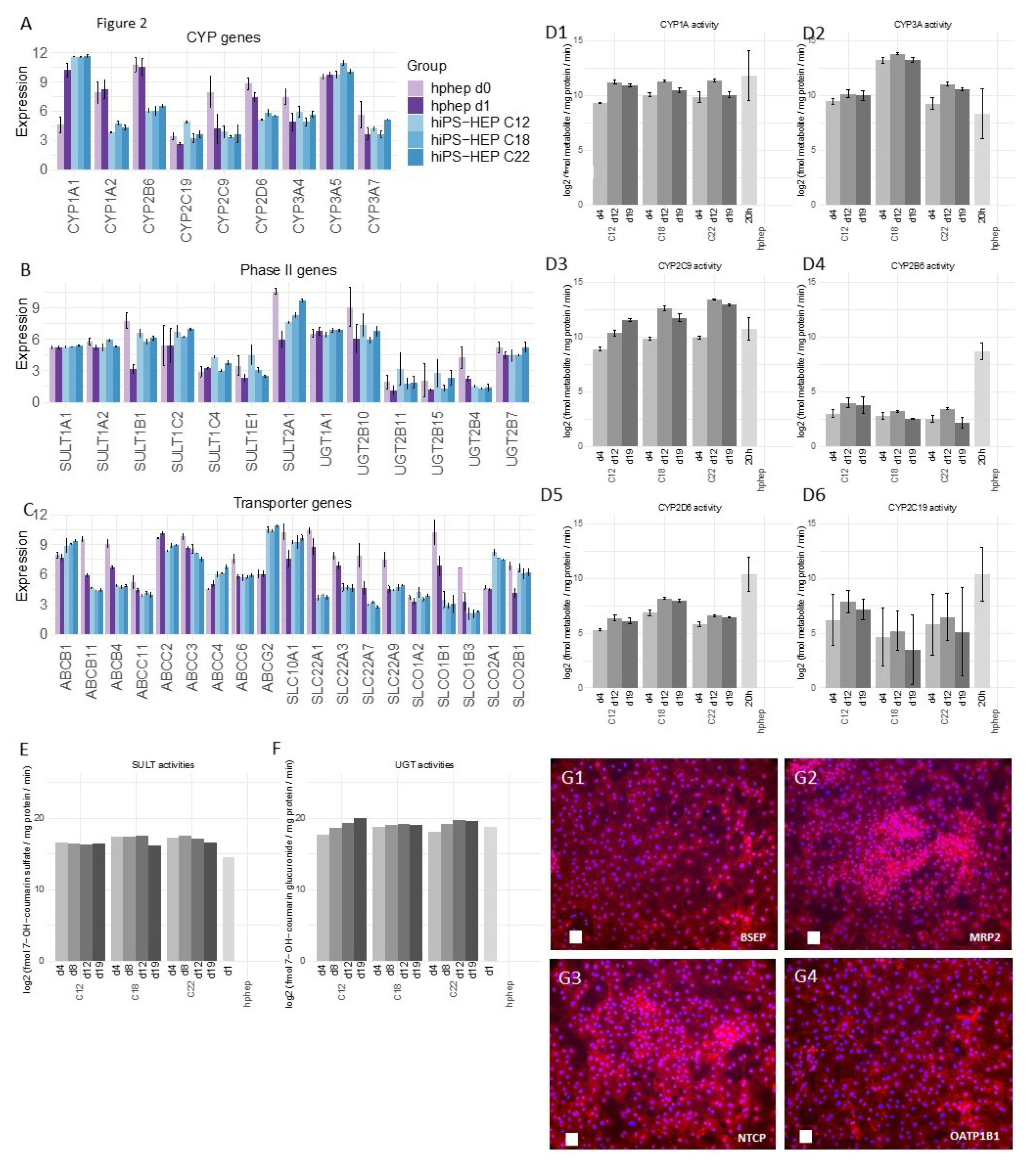
Furthermore, the reproducibility of the differentiation protocol is of great importance so that different cell batches show the same characteristics. We found that different hiPS-HEP batches derived from the same hiPSC line showed overall low variation measured as coefficient of variation (CV). For example, expression of hepatocyte markers (
Figure 1A) show CV < 5% for all genes except for
ASGR1 (CV < 8%), and expression of the drug metabolizing machinery show CV < 15% for the majority (87%) of all genes in
Figure 2A–C.
In contrast to the nearly uniform HNF4α-expression in hiPS-HEP, other markers or features are expressed only in a subpopulation of both hiPS-HEP and hphep, e.g., Albumin, α1-Antitrypsin, ASGPR1, and glycogen storage (
Figure 1 and
Figure 3). One would expect to find such different populations of hepatocytes since this is well-described for the liver and known as metabolic zonation [
10]. In the liver lobe, hepatocytes exist in periportal and perivenous phenotypes and therefore, it is important to look at features for both populations, e.g., albumin expression (predominantly in periportal hepatocytes) and CYP expression (predominantly in perivenous hepatocytes), when assessing a hepatocyte cell model. However, we cannot rule out that the observed heterogeneity for maturation markers such as Albumin and ASGPR1 in the hiPS-HEP cultures could be due to different levels of maturity.
We assessed the maturation status of the hiPS-HEP by performing transcriptomics analysis and functional assays in comparison to adult human primary hepatocytes. Albumin secretion, which is one hallmark for adult hepatocytes, was measured at comparable levels in hiPS-HEP derived from line ChiPSC12 and hphep (CV < 9%;
Figure 1D) and with slightly higher secretion in hiPS-HEP derived from line ChiPSC18 and ChiPSC22. This was confirmed by comparable
Albumin mRNA levels across both cell types (CV < 9%;
Figure 1A). Another characteristic of adult hepatocytes is high expression of the adult enzymes
CYP3A4 and low expression of the fetal enzyme
CYP3A7 which was the case in both cell types (
Figure 2A). ASGPR1, a marker for stem cell-derived hepatocytes enriched for primary hepatocyte features [
25], showed also comparable mRNA levels and immunostainings in hiPS-HEP and hphep (
Figure 1A,C3,C7) which suggested that hiPS-HEP have several adult hepatocyte features at levels comparable to hphep.
Two functionalities that require further improvement in hiPS-HEP are, (1) urea secretion and (2), the drug metabolizing machinery, comprising phase I and II enzymes as well as transporters. Urea secretion was clearly lower in hiPS-HEP than in hphep d1 (
Figure 1E). The only urea cycle gene that was lower in hiPS-HEP than in hphep was
ARG1 (
Figure 1B) which is the last in the chain of five key enzymes in the urea cycle [
26] and is likely the cause of the restricted urea production. Regarding phase I enzymes, the three main CYP enzymes, CYP1A, 3A, and 2C9, had comparable activity levels in hiPS-HEP and in hphep d1. CYP1A, 3A, and 2C9 combined metabolize about 60% of prescription drugs (CYP3A4 37%; CYP1A2 9%; CYP2C9 17%; [
27]), but in order to be useful for drug metabolism studies all enzymes need to be expressed at useful levels. Therefore, expression of CYP2B6, 2D6, and 2C19 needs to be improved in hiPS-HEP (
Figure 2D). We observed a similar situation for transporters as for the CYP enzymes: some were well expressed, e.g., MRP2 (
ABCC2) and NTCP (SLC10A1) and some need to be increased in hiPS-HEP, e.g.,
OATP1B1 and
OATP1B3 (
Figure 2C). In addition, formation of bile canaliculi was not observed, meaning that hiPS-HEP do not develop cell polarity with apical and basolateral membrane compartments. This limits the utility of the cells for applications requiring bile canaliculi formation, e.g., biliary excretion of drugs. Regarding phase II metabolism, we focused on UGTs and SULTs since they are responsible for more than half of all phase II metabolism of clinically used drugs [
28] and found SULT and UGT activity levels to be comparable in hiPS-HEP and hphep (
Figure 2E,F).
Noteworthy, Albumin and urea secretion as well as CYP and phase II activities were stable over time in hiPS-HEP (
Figure 1D–E;
Figure 2D–F). This phenotypic stability of the hiPS-HEP is clearly a superior feature over hphep, which are known to quickly loose functions such as CYP activities in conventional 2D cultures [
29,
30]. Many studies, for example for hepatitis virus studies and toxicity studies with repeated dosing, require a chronic treatment and a culture period of at least 2 weeks which is why phenotypic stability is of great importance for an in vitro hepatocyte model.
As expected, hphep display large variations of CYP activities between donors (see error bars for hphep in
Figure 2D) which reflect the large inter-individual variation existing in the population (see e.g., [
17]). This variation is a major issue that researchers are facing when using primary hepatocytes for in vitro models since the amount of hphep from one donor is limited and researchers are therefore forced to switch to new donors that may have a very different phenotype potentially affecting their studies. Therefore, one main advantage of hiPS-HEP is that one can obtain virtually unlimited amounts of cells from one donor, and importantly, there is only very low variation between different batches of hiPS-HEP, as seen for example in the reproducible and robust expression of hepatocyte markers (
Figure 1A), drug metabolizing genes (
Figure 2A–C), CYP activities (
Figure 2D), and the response to insulin (
Figure 3D). Noteworthy, inter-individual variation in the CYP activity profile can be observed when comparing hiPS-HEP derived from different iPSC lines: e.g., hiPS-HEP derived from ChiPSC18 have higher CYP3A and 2D6 activities than hiPS-HEP derived from the other 2 hiPSC lines (
Figure 2D2,D5), which is likely due to higher expression of the polymorphic genes CYP3A5 and 2D6 in ChiPSC18 (
Figure 2A).
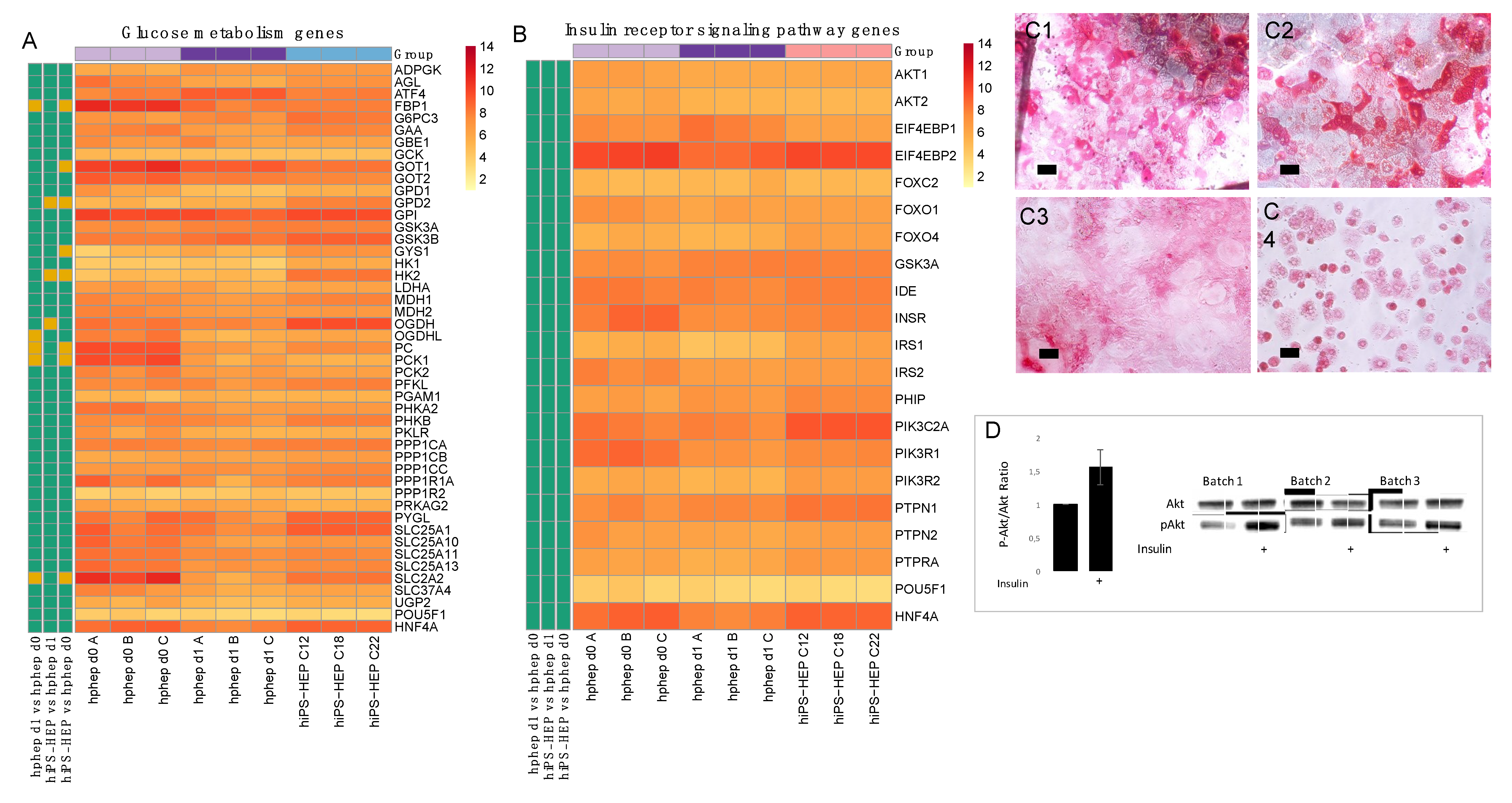
Energy metabolism is another key function of the liver. The results presented here indicate that hiPS-HEP have a comparable glucose and lipid metabolism as hphep, the gold standard (
Figure 3 and
Figure 4). However, several genes related to gluconeogenesis, e.g.,
PC, PCK1, FBP1, GOT1, GPD2, and
OGDH, are expressed on lower levels in hiPS-HEP (
Figure 3A). A potential cause could be the relatively high glucose concentration in the culture medium which would also explain the downregulation of gluconeogenesis-related genes in hphep d1 compared to hphep d0 (
Figure 3A).
One important area of use for hepatocytes is to model NAFLD, a common cause for chronic liver disease with a prevalence of 20–30% in Europe [
31]. NAFLD can progress to NASH which is associated with a higher risk of liver-related mortality and cardiovascular disease [
32], and further to cirrhosis with an increased risk for hepatocellular carcinoma [
33]. The long progression until clinical signs, the lack of good diagnostic tools as well as the absence of relevant preclinical models hampers the development of adequate treatments and drugs for NADFL/NASH, and highlights the need for novel translational in vitro models [
34]. Several animal models have been developed, however, due to the heterogeneous pathology few or none of these models represent the human situation accurately [
35]. Therefore, there is great and urgent need for an in vitro model for NAFLD/NASH.
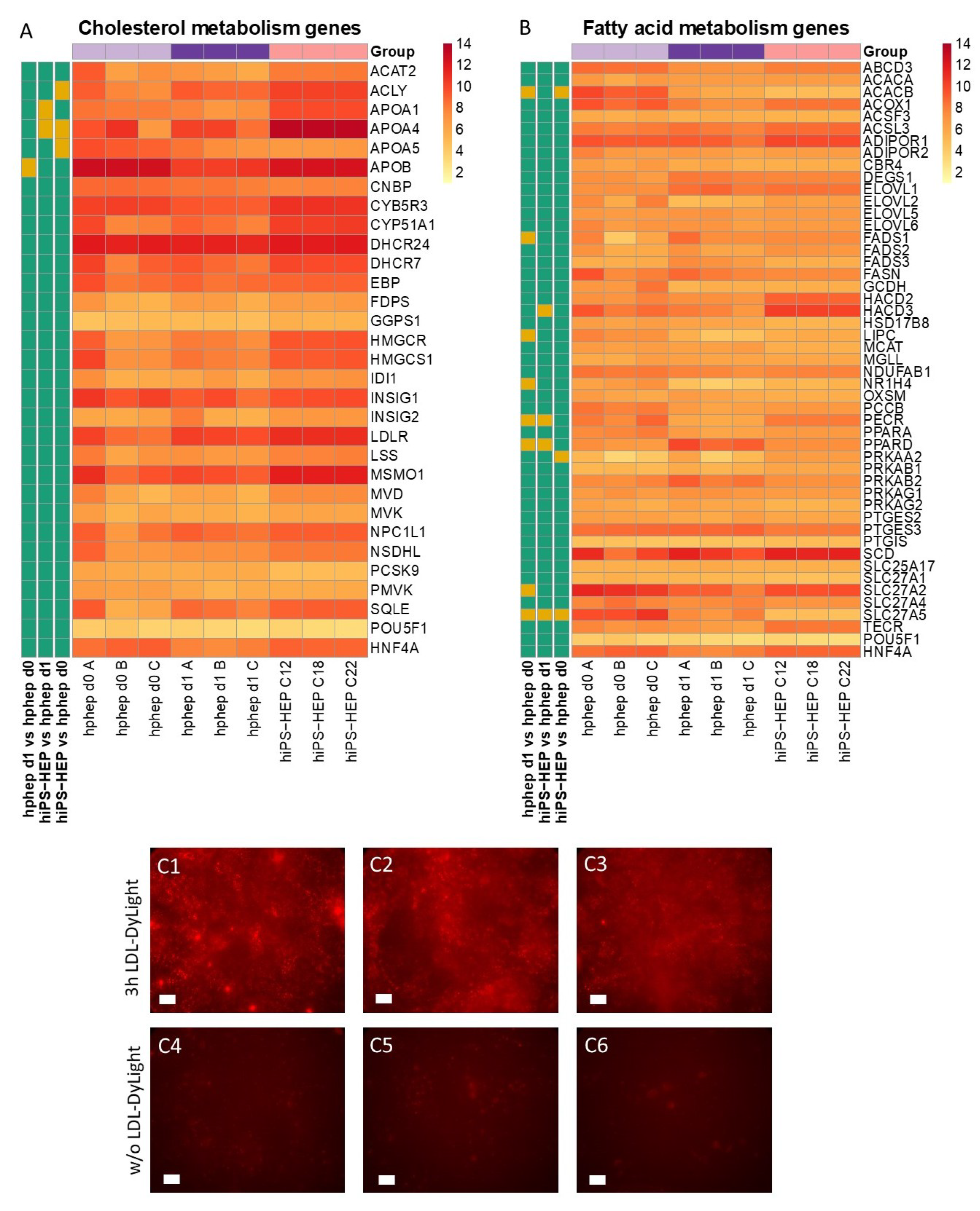
Our data presented here suggest that hiPS-HEP could serve as such a potential NAFLD/NASH model. Most genes related to lipid and cholesterol metabolism are expressed at comparable levels as in hphep d0/1 (
Figure 4). Only four genes differed significantly between hiPS-HEP and hphep d0/1, for example
APOA5, which is involved in lipoprotein metabolism by interacting with LDL receptor [
36]. However, despite the lower
APOA5 expression in hiPS-HEP and in agreement with substantial expression of the
LDL receptor, we found that hiPS-HEP accumulated labelled LDL upon a short incubation (
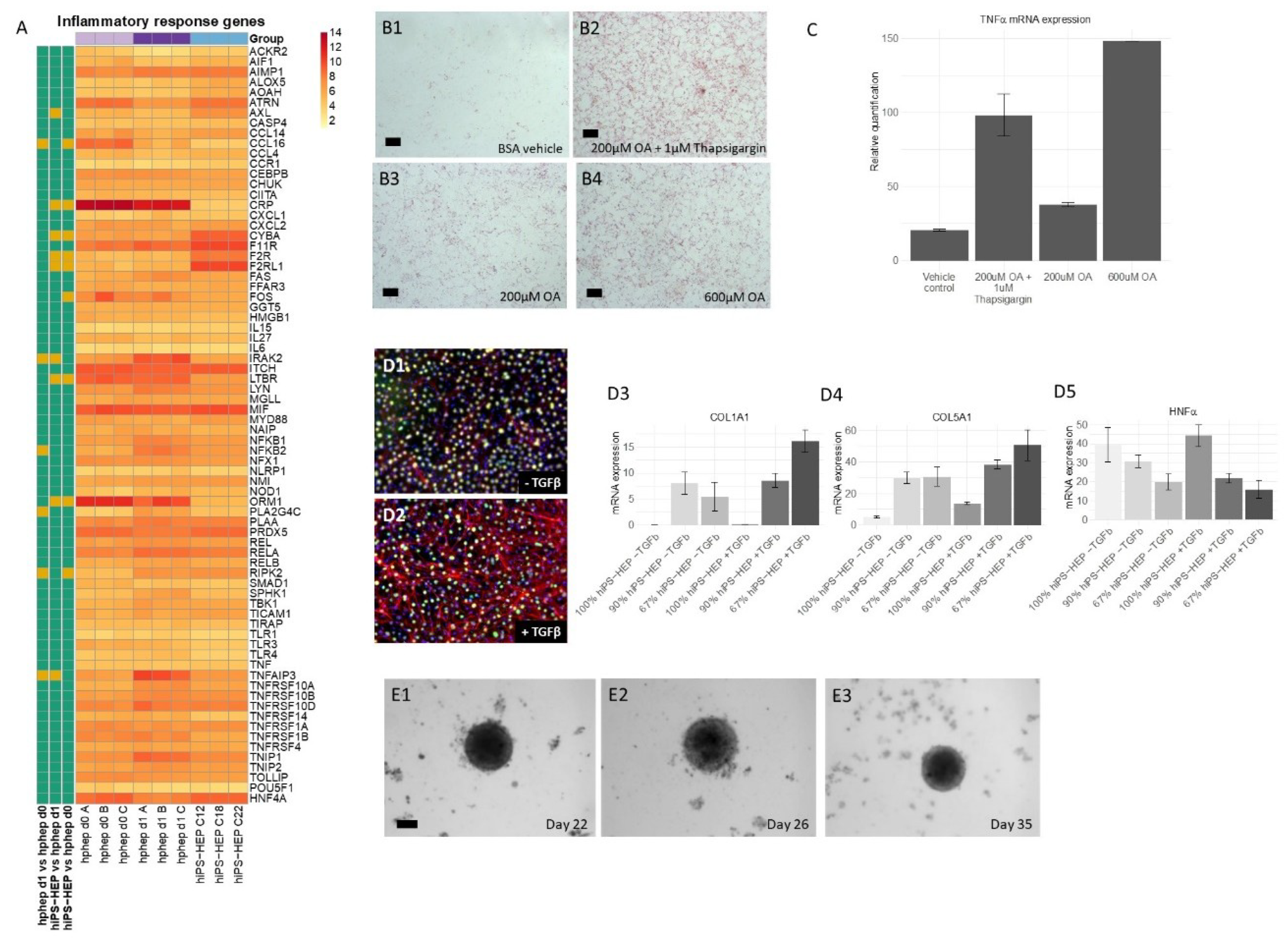
Figure 4A,C). We also observed dose-dependent steatosis, which in turn lead to dose-dependent upregulation of the inflammatory marker
TNFα (
Figure 5B,C). Importantly, under standard culture conditions most inflammation related genes were expressed at low levels both in hphep and hiPS-HEP (
Figure 5A), indicating a healthy status of the cells and no activation of an inflammatory response which is a prerequisite for the utility of the hiPS-HEP for modelling inflammation. Furthermore, hiPS-HEP responded in the anticipated manner to Thapsigargin-induced ER stress [
12,
37], with both an increased lipid accumulation and an upregulated
TNFα expression (
Figure 5B,C). Taken together, these results indicate that hiPS-HEP are comparable to adult primary hepatocytes regarding glucose and lipid metabolism as well as inflammatory markers. The observed responses are essential for a relevant human in vitro model for NAFLD/NASH and highlight the potential use of hiPS-HEP for such models. Another important finding is that key drug targets for NAFLD/NASH treatment are well expressed in hiPS-HEP, e.g., the peroxisome proliferator−activated receptor-α (
PPARA) and -δ (
PPARD), which are targets for the drug elafibranor [
38], and the nuclear receptor FXR (
NR1H4), which is a target for the drug obeticholic acid [
39]. All three genes are expressed on the same level in hiPS-HEP and hphep d0 (
Figure 4B) which demonstrates the potential of hiPS-HEP for drug target studies for NAFLD/NASH.
Since the progression of NAFLD to NASH includes a contribution of inflammatory and fibrogenic response from non-parenchymal cells, hepatocytes need to be co-cultured with non-parenchymal cells, such as stellate cells and Kupffer cells, as reviewed in [
35]. To date, only a limited number of attempts using primary cells or cell lines (reviewed in [
40]) or hepatocytes derived from human pluripotent stem cells [
12,
41] have been reported in the NASH field. However, in the two NASH studies utilizing stem cell-derived hepatocytes, those were not co-cultured with non-parenchymal cells [
12,
41]. Two other studies described 2D co-cultures of several hiPSC-derived liver cell types: liver progenitor cells and HSC [
42], and liver progenitor cells, liver sinusoidal endothelial cells and HSC [
43], respectively, and reported a positive effect of the co-culture on the maturity of the hiPSC-liver progenitor cells but did not evaluate the utility of the co-cultures for modeling NASH. Another study reported modeling of liver fibrosis using the hepatoma cell line HepaRG and hiPSC-derived HSC [
44]. To the best of our knowledge, our study is the first one to describe co-cultures of hiPSC-derived hepatocytes and human primary HSC. Importantly, we found that α-SMA expression, a hallmark of activated HSC and fibrosis, was induced in HSC in 2D co-cultures by TGFβ treatment. Since HSC showed a weak activation in 2D even without TGFβ stimulation, we generated 3D-spheroids of hiPS-HEP and HSC which can be maintained for at least 35 days. The results presented here pave the way for further exploring the utility of these co-culture spheroids for NASH studies. The 3D co-culture setting may even further improve the hiPS-HEP functionality compared to 2D cultures, similarly to reports on 3D spheroids consisting of hphep and non-parenchymal cells [
8].
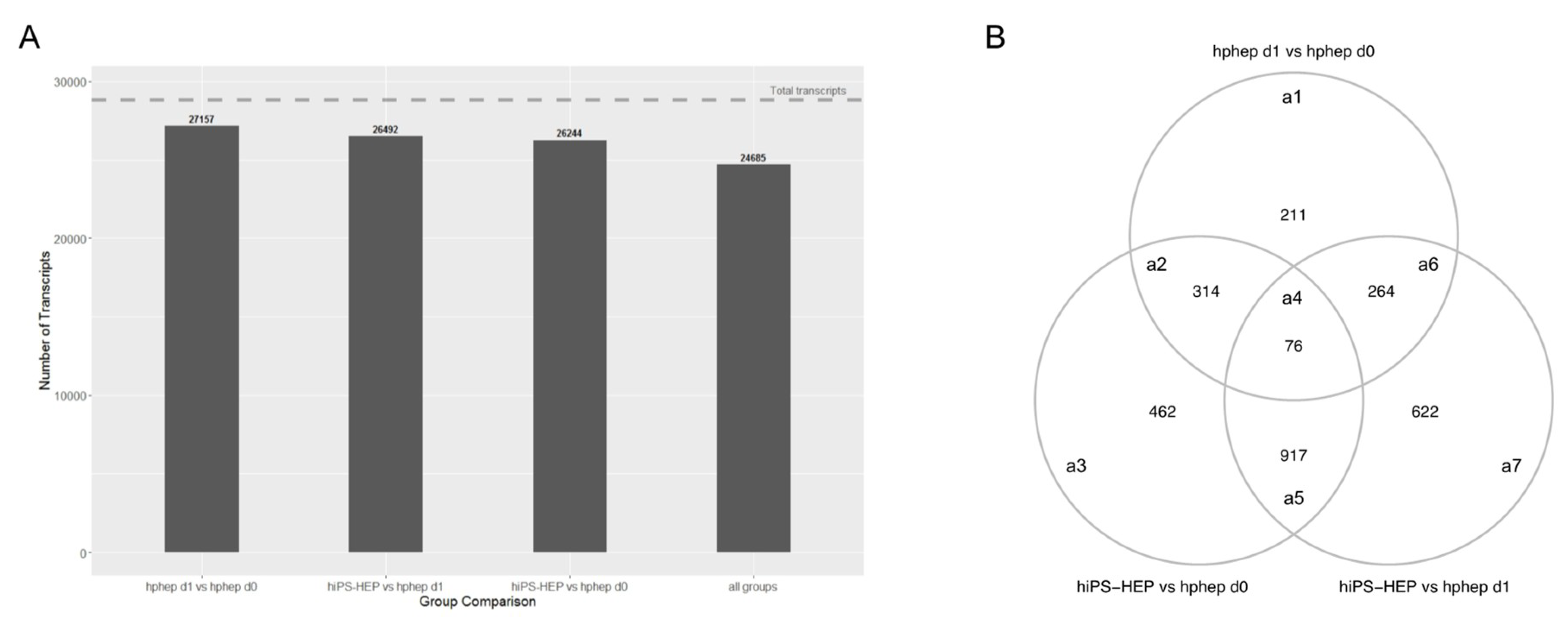
The global analysis of the transcriptomics data revealed very similar phenotypes of the three cell populations (
Figure 6A). Differences were observed, amongst others, in phase I and II metabolism, indicating a lower functionality of hiPS-HEP and hphep d1 compared to hphep d0, which is in line with our gene expression and functional data on drug metabolism (
Figure 2). Interestingly, in the DEGs between hiPS-HEP and hphep d0 and d1 “Attenuation phase” and “HSP1-dependent transactivation” are over-represented pathways which are related to cellular response to heat stress. It has been reported previously that cryopreservation and re-plating of hphep results in reduced ability to form cell-matrix and cell-cell interactions which triggers stress and heat shock responses [
45]. Our observation that DEGs involved in these pathways (
HSPA1A, HSPA1B, and
DNAJB1) are upregulated in hphep d0 and d1 compared to hiPS-HEP is in line with these reports. Furthermore, pathways connected to ECM and laminin interactions are over-represented in the DEGs between hiPS-HEP and hphep d0 and d1, with higher expression in hiPS-HEP. A possible explanation could be that hiPS-HEP are differentiated in 2D cultures whereas hphep come from a 3D environment. It would be interesting to investigate if these pathways are downregulated in hiPS-HEP 3D spheroids.
The identification of DEGs is of great importance since they can provide a basis for future improvements as the aim is to reach the same functionality and maturity in hiPS-HEP as in hphep d0. One incentive for such continuous efforts is that hphep are not useful as a cell source for gene editing and are limited for disease modeling of a specific genetic background. The advance of the hiPSC technology allows to routinely generate hiPSC lines from many individuals, including healthy persons and patients, and thus represent the whole population. Furthermore, genetic modification of hiPSC lines using for example CRISPR/Cas9, allows to study the molecular basis of diseases.
Taken together, the hiPS-HEP used in this study represent a homogenous cell population with a high similarity to hphep and a potential for modeling NAFLD/NASH. Our results highlight the potential of hiPSC-derived hepatocytes as a virtually unlimited cell source. In order to build more advanced cell models, to better recapitulate the human liver environment, to reflect the population diversity and to increase the predictability for disease modeling, advanced 3D culture systems including multiple cell types and microfluidics as well as panels of hiPSC lines derived from various ethnicities need to be developed and implemented.
4. Materials and Methods
4.1. Cell Culture
Cryopreserved hiPS-HEPs derived from the hiPSC lines ChiPSC12, ChiPSC18, and ChiPSC22 were thawed, plated, and maintained according to the vendor instructions (Cellartis Enhanced hiPS-HEP v2 kits, Cat.no. Y10133, Y10134, Y10135, Takara Bio Europe, Gothenburg, Sweden,
www.takarabio.com). Enhanced hiPS-HEP v2 were maintained for up to 21 days post-thawing with media changes every 2 or 3 days using the Cellartis Enhanced hiPS-HEP Long-Term Maintenance Medium included in the kits.
For 2D co-cultures of Enhanced hiPS-HEP v2 and primary human stellate cells (HCS, BioIVT, Brussels, Belgium), cells were seeded on a Fibronectin coating (concentration 2 µg/cm2; Roche, Basel, Swiss) in a 2:1 ratio (hiPS-HEP: HSC) at a cell density of 400 K/cm2 using the regular plating medium included in the hiPS-HEP v2 kits. Instead of the regular HEP Long-Term Maintenance Medium the following maintenance medium was used for the co-cultures: Williams’ Medium E supplemented with 0.1% PEST (15140–130, ThermoFisher, Stockholm, Sweden), 0.5% DMSO (D2650, Sigma, Darmstadt, Germany), 0.67% Cellartis Hepatocyte supplement (Y11149, Takara Bio), 2% Cellartis Hepatocyte Additives (Y11078, Takara Bio), 194 µM L-Proline (P5607, Sigma), and 173 µM L-ascorbic acid 2-phosphate sesquimagnesium salt (A8960, Sigma).
For 3D co-cultures of Enhanced hiPS-HEP v2 and HSC, cells were seeded in a 2:1 ratio (hiPS-HEP: HSC) in InVitroGRO CP medium (BioIVT) supplemented with 0.5% PEST, and 2% Cellartis Hepatocyte Additives (Y11078, Takara Bio) in Costar 96 well ultra-low attachment plates (7007, Corning, Amsterdam, The Netherlands) with 30,000 cells per well in 100 µL plating medium. At 24 h post-seeding, medium was changed to 3D spheroid maintenance medium as described by Rashidi and colleagues [
46]. At 24–48 h post-seeding, spheroids had formed and were maintained for up to 35 days with 50% medium changes every 2–3 days. Alternatively, spheroids could also be maintained with the 2D co-culture medium described above.
Cryopreserved hphep from three donors (Cat.no. M00995-P for male donors, F00995-P for female donors, BioIVT), selected based on high viability (see
Table 1 for donor details), were thawed and plated according to the provider’s instructions. For experiments that required the hepatocytes to be cultured for 24 h, medium was changed 4 h post-plating to fresh plating medium (InVitroGRO CP medium, BioIVT).
4.2. Transcriptomics Analysis
4.2.1. Total RNA Extraction
Two batches of Enhanced hiPS-HEP v2 from each hiPSC line (ChiPSC12, ChiPSC18, and ChiPSC22) were harvested on day 13 post-thawing by scraping the cells in the culture medium, centrifuging at 300×
g for 5 min and frozen as dry cell pellets. Human primary hepatocytes (
n = 3 donors) were harvested as dry cell pellets directly after thawing (day 0) and on day 1 post-thawing (day 1). Cell pellets were stored at −80 °C until RNA extraction. Total RNA was extracted from the cell pellets using the GenElute RNA/DNA/protein Plus Purification kit (E5163, Sigma Aldrich). RNA amounts were quantified using a NanoDrop ND-1000 (NanoDrop,
http://www.nanodrop.com).
4.2.2. Gene Array
The quality of the RNA and cDNA was verified using a 2100 Agilent Bioanalyzer. To measure the mRNA levels, cDNA was synthesized from the RNA samples applying the GeneChip WT PLUS Reagent Kit (Affymetrix, Stockholm, Sweden) and fragmented cDNA was hybridized at 45° Celsius for 16 h to whole transcript HuGene ST 2.0 arrays (Affymetrix,
http://www.affymetrix.com) at SCIBLU Genomics (Lund University, Sweden). In total, 12 expression microarrays were run.
4.2.3. Data Processing
Raw microarray data was imported into R (version 3.6.1, R Core Team, 2019,
https://www.r-project.org/) and signal intensities normalized by the Robust Multichip Average method in the oligo package [
47]. The two biological replicates for each hiPSC line were summarized by mean expression. To remove transcripts with expression values close to background, probes with a log
2 expression below 5 in all samples were removed. The resulting dataset contained 21,427 transcripts and 9 samples (3 hphep day 0, 3 hphep day 1, 3 hiPS-HEP). The transcripts were mapped to Human Genome Organization Gene Nomenclature Committee (HGNC) symbols using the HuGene 2.0 ST V1 NetAffx file from Affymetrix (NA36, genome build hg19,
http://www.affymetrix.com/analysis/index.affx). The microarray data used in this study follow the MIAME standard and raw expression data are available at ArrayExpress (
https://www.ebi.ac.uk/arrayexpress/), accession number E-MTAB-8286.
4.2.4. Statistical Analysis
Inspection of transcriptomics data revealed an approximate gamma distribution, and thus statistical testing for differential expression was based on a generalized linear model (GLM) from the gamma family with a linear link. The GLM was fitted using genes as response variables and sample group (hphep day 0, hphep day 1, hiPS-HEP) as covariates. The primary human hepatocytes day 0 and day 1 samples were treated as paired. Statistical significance of differential expression was assessed with the likelihood ratio test. p-values were adjusted for multiple testing by Benjamini-Hochberg correction. Differentially expressed genes were identified using a combined criterion of adjusted p-value < 0.05 and absolute log2 fold change > 2.
Pathway over-representation analysis of the differentially expressed genes was carried out with Reactome Pathway Database [
48,
49], using a criterion of a
p-value < 0.05, and at least two differentially expressed genes present for the identified over-represented pathways.
4.3. Immunocytochemistry
Cells were stained as described previously in [
7]. Briefly, Enhanced hiPS-HEP v2 were fixed on day 12 post-thawing and hphep on day 1 post-thawing, by 15 min incubation with 4% Formaldehyde. Cells were stained with the following primary and secondary antibodies: rabbit anti-α1AT (A0012, DAKO, 1:200 dilution), rabbit anti-albumin (A0001, DAKO, 1:1000), mouse anti-ASGPR1 (MAI-40244, ThermoFisher 1:50), rabbit-anti-BSEP (purchased from Bruno Stieger, University Hospital Zurich, Zurich, Switzerland, 1:100); rabbit anti-HNF4α (SC-8987, SantaCruz Biotechnology, 1:300, Heidelberg, Germany), rabbit anti-MRP2 (SC-20766, SantaCruz Biotechnology, 1:50), rabbit anti-NTCP (Bruno Stieger, 1:400), rabbit anti-OATP1B1 (Bruno Stieger, 1:200), donkey anti-rabbit Alexa 594 IgG (A21207, ThermoFisher, 1:1000) or goat anti-mouse Alexa 488 (A11029, ThermoFisher, 1:1000). For nuclear counterstaining, DAPI (Sigma, D9542) was added during the incubation with the secondary antibodies (add 2 µL/mL of a 1 mg/mL DAPI stock in DMSO). Stainings were examined using an inverted fluorescence microscope (Eclipse Ti-U, Nikon, Amsterdam, The Netherlands), ANDOR Zyla sCMOS digital camera and the NIS-Elements software package (version 4.30). Technical control staining without primary antibodies were performed for all secondary antibodies and these were negative. Quantification of HNF4α positive cells was done in relation to DAPI stained cells using the CellC Cell Counting Software [
50].
Co-cultures of Enhanced hiPS-HEP v2 and HSC were permeabilized with 0.5% Triton X-100 solution in PBS for 1 h at room temperature (RT) and then incubated for 30 min at RT with blocking buffer containing 1% BSA and 10% normal goat serum (Sigma). Primary and secondary antibodies were diluted in blocking buffer. Cells were incubated for 2 h at RT (α-SMA) or overnight at 4 °C (HNF4α) with the primary antibodies and subsequently washed three times with PBS before incubating with the secondary antibodies for 1 h at RT. Next, cells were washed three times with PBS, stained for 5 min with DAPI diluted 1:25,000 in PBS and washed again. The following primary and secondary antibodies were used: mouse anti-α-smooth muscle actin (α-SMA; 1:1000; A5228, Sigma), mouse anti-HNF4α (1:200; ab41898, Abcam), goat anti-mouse Alexa Fluor 594 (1:300; ThermoFisher Scientific), and goat anti-mouse FITC (1:50; ThermoFisher Scientific). Fluorescent images were acquired on an Axio Observer Z1 Zeiss microscope (Zeiss, Breda, The Netherlands) and processed with ZEN 2.3 lite software (Zeiss).
4.4. Period Acid-Schiff (PAS) Staining
Glycogen storage was visualized by PAS staining of Enhanced hiPS-HEP v2 derived from C12, C18, and C22 (fixed on day 12 post-thawing) and hphep (fixed on day 1 post-thawing). Cells were stained with the glycogen assay kit (MAK016, Sigma) according to the manufacturer’s protocol.
4.5. Albumin Secretion
Albumin secretion from Enhanced hiPS-HEP v2 (derived from C12, C18, and C22) was analyzed on days 4, 6, 12, and 20 post-thawing and from hphep cells 24 h post-thawing. The culture medium was collected after 24 h of conditioning and Albumin content was analyzed with the Albuwell kit (Exocell, Philadelphia, PA) according to the manufacturer’s protocol. The Albumin content in the medium was normalized to the assay duration (24 h) and the amount of protein per well.
For protein quantification, cells were washed once with DPBS (with Calcium and Magnesium) and lysed in 0.02 mM NaOH over night at 4 °C and stored at −20 °C until quantification using the Pierce BCA Protein Assay kit (ThermoFisher, Rockford, IL) according to the manufacture’s instruction.
4.6. Urea Secretion
On days 13 and 20 post-thawing, Enhanced hiPS-HEP v2 and hphep d1 were incubated with 5 mM ammonium chloride for 24 h. After 24 h, medium was collected, and urea secretion was analyzed with the QuantiChrom Urea Assay Kit (BioAssay Systems, Hayward, CA, USA). Urea content was normalized to the amount of protein per well (determined using the Pierce BCA Protein Assay Kit, see above) and the assay duration (24 h).
4.7. AKT Western Blot
Phosphorylated AKT: Enhanced hiPS-HEP (derived from C18, on day 12 post-thawing) were incubated in insulin-free medium (phenol-red free Williams’ medium E containing 0.1% PEST, 25 mM HEPES, 2 mM L-Glutamine) for 3 h and then treated for 10 min with 0 nM or 100 nM insulin. Phosphorylated AKT (Cell Signaling) and total AKT (Cell Signaling) were quantified by Western blot (NuPAGE 4–12% Bis-Tris Protein Gels, Thermo Fisher Scientific).
4.8. CYP Activity Assay
The CYP activities of Enhanced hiPS-HEP v2 were analyzed by performing a CYP activity assay at days 4, 12, and 19 after thawing and the results were compared to hphep cultured for 20 h. Briefly, the cells were carefully washed twice with pre-warmed Williams’ medium E (Phenol-red free, +0.1% PEST). Then, the activity assay was started by adding 110 μL/cm
2 culture area of pre-warmed Williams’ medium E (phenol-red free) containing 0.1% PEST, 25 mM HEPES (H7523, Sigma), 2 mM L-Glutamine, and the probe substrate cocktail (see
Table 2 below). After a 2-h incubation at 37 °C, 100 μL of the supernatant was collected and kept at −80 °C until LC/MS analysis. LC/MS analysis (performed at Pharmacelsus GmbH, Saarbrücken, Germany) was used to measure the formation of the specific metabolites Acetaminophen (CYP1A), OH-Bupropion (CYP2B6), 4-OH-Diclofenac (CYP2C9), 4-OH-Mephenytoin (CYP2C19), OH-Bufuralol (CYP2D6), and 1-OH-Midazolam (CYP3A). The metabolite concentrations were normalized to the amount of protein per well (determined using the Pierce BCA Protein Assay Kit, see above) and the assay duration (120 min). To be able to normalize the results between different LC/MS runs, a metabolite cocktail with known concentrations of all metabolites is included in every analysis batch.
4.9. Phase II Enzyme Activity Assay
The phase II enzyme activities of Enhanced hiPS-HEP v2 were analyzed day 4, 8, 12, and 19 after thawing by performing a phase II activity assay. Briefly, the cells were carefully washed twice with pre-warmed Williams’ medium E (+0.1% PEST). Then the activity assay was started by adding 110 μL/cm2 culture area of pre-warmed Williams’ medium E containing 0.1% PEST, 25 mM HEPES, 2 mM L-Glutamine, and 200 μM 7-OH-coumarin. After a 2 h incubation at 37 °C, 100 μL of the supernatant was collected and kept at −80 °C until LC/MS analysis. LC/MS analysis (performed at Pharmacelsus GmbH) was used to measure the formation of 7-OH-coumarin sulfate and 7-OH-coumarin glucuronide, specific metabolites for sulfotransferases and UDP-glucuronosyltransferases, respectively. The metabolite concentrations were normalized to the amount of protein per well (determined using the Pierce BCA Protein Assay Kit, see above) and the assay duration (120 min).
4.10. LDL Uptake
In order to determine the uptake of low-density lipoproteins (LDL), Enhanced hiPS-HEP v2 (derived from C12, C18, and C22), on day 6 after thawing, were incubated for 3 h with LDL-DyLight (Cat.no. 10011125, Cayman Chemical, Hamburg, Germany) diluted 1:100 in regular maintenance medium. Next, cells were washed once with DPBS (with Calcium and Magnesium) and immunofluorescence was recorded as described above under immunocytochemistry.
4.11. Fatty Acid Accumulation and Inflammatory Response
Enhanced hiPS-HEP v2 were incubated for 24 h (on days 5–6 post-thawing) with Williams’ medium E containing 0.1% PEST, 25 mM HEPES, and 2 mM L-Glutamine supplemented with either 200 µM oleic acid (OA, O1008, Sigma) coupled to 77 µM fatty acid-free BSA (FAF-BSA, A8806, Sigma), 600 µM OA coupled to 231 µM FAF-BSA, or only 231 µM FAF-BSA (as vehicle control). In addition, one group of cells was treated with 200 µM OA (coupled to 77 µM FAF-BSA) plus 1 µM Thapsigargin (T0933, Sigma). After the 24 h incubation, cells were either stained with Oil Red O or harvested for gene expression analysis. Oil Red O staining was performed using the Hepatic Lipid Accumulation/Steatosis Assay Kit (Cat.no. ab133131, Abcam) according to the protocol provided with the kit. Stainings were evaluated using a Zeiss AxioVert microscope, an Axicam 105 color camera and the ZEN2 software (all from Carl Zeiss, Jena, Germany). For gene expression analysis, cells were harvested in RNAprotect Cell Reagent (Cat No. 76526, Qiagen, Hilden, Germany). RNA preparation, cDNA synthesis, and qPCR were performed as described previously in [
7]. Gene expression was analyzed using the TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA):
TNFα (Hs00174128_m1), and
CEBPα (Hs00269972_s1) which served as a reference gene.
4.12. RNA Prep and RT-qPCR of Co-Cultures and 3D Spheroids
Total RNA from co-cultures and from 3D spheroids was isolated using the Ambion RNAqueous RNA Isolation Kit (ThermoFisher). The manufacturer’s supplied protocol was followed during the isolation and RNA concentrations were measured using the nanodrop ND-1000 spectrophotometer. Cellular RNA (maximum of 200 ng) was reverse transcribed into cDNA using the High Capacity RNA to cDNA kit (Applied Biosystems) in a total reaction volume of 20 µL consisting of 9 µL assay buffer, 1 µL enzyme mixture and added up to 20 µL with RNAse free H2O. Samples were incubated for 60 min at 37 °C and 5 min at 95 °C using a BioRad iCycler. qPCR samples were subsequently prepared by diluting cDNA samples 1:1 with RNAse free H2O and samples consisted of 2 µL diluted cDNA, 0.625 µL assay on demand (AoD, ThermoFisher) or 25 µM primers with 6.25 µL Taqman® Gene expression mastermix (ThermoFisher) filled up to a total volume of 12.5 µL with RNAse free H2O. Samples were run at 50 °C for 2 min, 95 °C for 10 min and 40–45 cycles, each cycle consisting of 95 °C for 15 s and 60 °C for 1 min using the QuantStudio6 flex. The following TaqMan Gene Expression Assays were used: Collagen 1a1 (Hs01076777_m1), Collagen 5a1 (Hs00609133_m1), HNF4α (Hs00230853_m1), and HPRT1 (Hs03929098_m1). The latter served as a reference gene.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}