Genetically Engineered Pigs to Study Cancer

Abstract
:
1. Introduction
2. Large Mammals as Biomedical Models
3. Generation of Genetically Modified Pigs
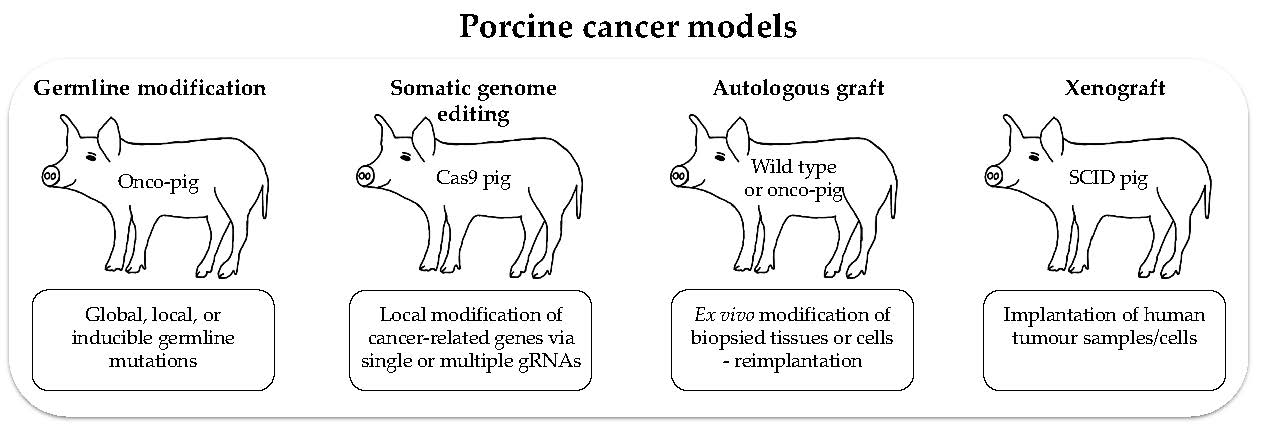
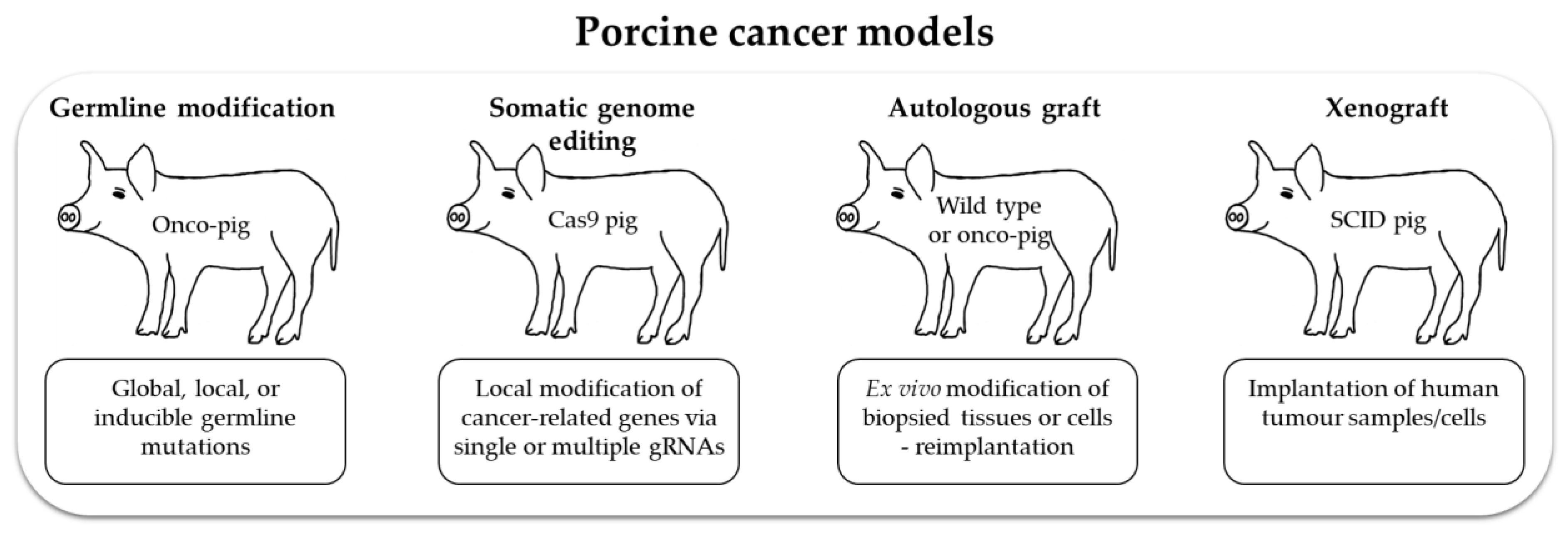
4. Porcine Cancer Models
4.1. Porcine Models for Breast Cancer
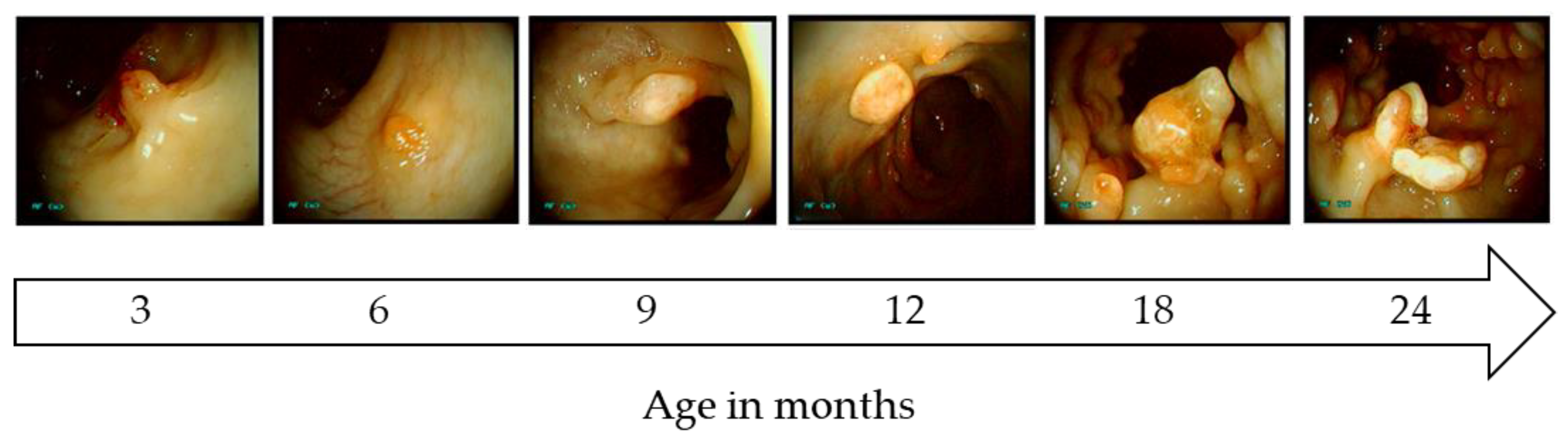
4.2. Porcine Models for Colorectal Cancer
4.3. Porcine Models for Pancreatic Cancer
4.4. Porcine Models for Osteosarcoma
5. Porcine Tumor Xenograft Models
6. Future Perspectives and Challenges
Author Contributions
Funding
Conflicts of Interest
References
- Cosco, T.D.; Howse, K.; Brayne, C. Healthy ageing, resilience and wellbeing. Epidemiol. Psychiatr. Sci. 2017, 26, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Jaul, E.; Barron, J. Age-Related Diseases and Clinical and Public Health Implications for the 85 Years Old and Over Population. Front. Public Health 2017, 5, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; Anderson, B.O.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Hayes, M.N.; Moore, F.E.; Tang, Q.; Garcia, S.P.; Blackburn, P.R.; Baxi, K.; Wang, L.; Jin, A.; Ramakrishnan, A.; et al. tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. eLife 2018, 7. [Google Scholar] [CrossRef]
- Park, J.T.; Leach, S.D. Zebrafish model of KRAS-initiated pancreatic cancer. Anim. Cells Syst. 2018, 22, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Leng, X.; Wang, G.; Wan, X.; Cao, H. The construction of intrahepatic cholangiocarcinoma model in zebrafish. Sci. Rep. 2017, 7, 13419. [Google Scholar] [CrossRef] [Green Version]
- White, R.M.; Sessa, A.; Burke, C.; Bowman, T.; LeBlanc, J.; Ceol, C.; Bourque, C.; Dovey, M.; Goessling, W.; Burns, C.E.; et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008, 2, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Antinucci, P.; Hindges, R. A crystal-clear zebrafish for in vivo imaging. Sci. Rep. 2016, 6, 29490. [Google Scholar] [CrossRef] [Green Version]
- Nicoli, S.; Ribatti, D.; Cotelli, F.; Presta, M. Mammalian tumor xenografts induce neovascularization in zebrafish embryos. Cancer Res. 2007, 67, 2927–2931. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cao, Z.; Zhang, X.M.; Nakamura, M.; Sun, M.; Hartman, J.; Harris, R.A.; Sun, Y.; Cao, Y. Novel mechanism of macrophage-mediated metastasis revealed in a zebrafish model of tumor development. Cancer Res. 2015, 75, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhang, Y.; Lim, S.; Hosaka, K.; Yang, Y.; Pavlova, T.; Alkasalias, T.; Hartman, J.; Jensen, L.; Xing, X.; et al. A Zebrafish Model Discovers a Novel Mechanism of Stromal Fibroblast-Mediated Cancer Metastasis. Clin. Cancer Res. 2017, 23, 4769–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Gao, B.; Zhang, W.; Qian, Z.; Xiang, Y. Monitoring antiangiogenesis of bevacizumab in zebrafish. Drug Des. Dev. Ther. 2018, 12, 2423–2430. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Wang, G.; Xiao, Q.; Zhou, Y.; Wei, Y.; Gong, Z. Antiangiogenic effects of AA-PMe on HUVECs in vitro and zebrafish in vivo. Oncotargets Ther. 2018, 11, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- Dolensek, J.; Rupnik, M.S.; Stozer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef] [Green Version]
- Steiniger, B.S. Human spleen microanatomy: Why mice do not suffice. Immunology 2015, 145, 334–346. [Google Scholar] [CrossRef]
- Holliday, R. Neoplastic transformation: The contrasting stability of human and mouse cells. Cancer Surv. 1996, 28, 103–115. [Google Scholar]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef]
- Watson, A.L.; Carlson, D.F.; Largaespada, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Engineered Swine Models of Cancer. Front. Genet. 2016, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Hagai, T.; Chen, X.; Miragaia, R.J.; Rostom, R.; Gomes, T.; Kunowska, N.; Henriksson, J.; Park, J.E.; Proserpio, V.; Donati, G.; et al. Gene expression variability across cells and species shapes innate immunity. Nature 2018, 563, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martignoni, M.; Groothuis, G.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Alvarez, C.E. Naturally occurring cancers in dogs: Insights for translational genetics and medicine. ILAR J. 2014, 55, 16–45. [Google Scholar] [CrossRef] [Green Version]
- MacEwen, E.G. Spontaneous tumors in dogs and cats: Models for the study of cancer biology and treatment. Cancer Metastasis Rev. 1990, 9, 125–136. [Google Scholar] [CrossRef]
- Perleberg, C.; Kind, A.; Schnieke, A. Genetically engineered pigs as models for human disease. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Flisikowska, T.; Kind, A.; Schnieke, A. Genetically modified pigs to model human diseases. J. Appl. Genet. 2014, 55, 53–64. [Google Scholar] [CrossRef]
- Lunney, J.K. Advances in swine biomedical model genomics. Int. J. Biol. Sci. 2007, 3, 179–184. [Google Scholar] [CrossRef]
- Hoffe, B.; Holahan, M.R. The Use of Pigs as a Translational Model for Studying Neurodegenerative Diseases. Front. Physiol. 2019, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Flisikowska, T.; Kind, A.; Schnieke, A. Pigs as models of human cancers. Theriogenology 2016, 86, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.J.; Kissinger, C.B.; McCain, R.R.; Cooper, B.R.; Marchant-Forde, J.N.; Vreeman, R.C.; Hannou, S.; Knipp, G.T. Assessment of juvenile pigs to serve as human pediatric surrogates for preclinical formulation pharmacokinetic testing. AAPS J. 2013, 15, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.J.; Farrell, D.E.; Howard, K.D.; Kawalek, J.C. Identification of multiple constitutive and inducible hepatic cytochrome P450 enzymes in market weight swine. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 908–915. [Google Scholar]
- Hammer, R.E.; Pursel, V.G.; Rexroad, C.E., Jr.; Wall, R.J.; Bolt, D.J.; Ebert, K.M.; Palmiter, R.D.; Brinster, R.L. Production of transgenic rabbits, sheep and pigs by microinjection. Nature 1985, 315, 680–683. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Lv, D.; Ma, T.; Deng, S.; Yang, M.; Song, Y.; Zhang, X.; Zhang, J.; Fu, J.; Lian, Z.; et al. AANAT transgenic sheep generated via OPS vitrified-microinjected pronuclear embryos and reproduction efficiency of the transgenic offspring. PeerJ 2018, 6, e5420. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512. [Google Scholar] [CrossRef]
- Nowak-Imialek, M.; Niemann, H. Pluripotent cells in farm animals: State of the art and future perspectives. Reprod. Fertil. Dev. 2012, 25, 103–128. [Google Scholar] [CrossRef]
- Blomberg, L.A.; Telugu, B.P. Twenty years of embryonic stem cell research in farm animals. Reprod. Domest. Anim. 2012, 47, 80–85. [Google Scholar] [CrossRef]
- Gao, X.; Nowak-Imialek, M.; Chen, X.; Chen, D.; Herrmann, D.; Ruan, D.; Chen, A.C.H.; Eckersley-Maslin, M.A.; Ahmad, S.; Lee, Y.L.; et al. Establishment of porcine and human expanded potential stem cells. Nat. Cell Biol. 2019, 21, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.H.; McWhir, J.; Ritchie, W.A.; Wilmut, I. Sheep cloned by nuclear transfer from a cultured cell line. Nature 1996, 380, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Schnieke, A.E.; Kind, A.J.; Ritchie, W.A.; Mycock, K.; Scott, A.R.; Ritchie, M.; Wilmut, I.; Colman, A.; Campbell, K.H. Human factor IX transgenic sheep produced by transfer of nuclei from transfected fetal fibroblasts. Science 1997, 278, 2130–2133. [Google Scholar] [CrossRef] [PubMed]
- McCreath, K.J.; Howcroft, J.; Campbell, K.H.; Colman, A.; Schnieke, A.E.; Kind, A.J. Production of gene-targeted sheep by nuclear transfer from cultured somatic cells. Nature 2000, 405, 1066–1069. [Google Scholar] [CrossRef]
- Park, K.W.; Cheong, H.T.; Lai, L.; Im, G.S.; Kuhholzer, B.; Bonk, A.; Samuel, M.; Rieke, A.; Day, B.N.; Murphy, C.N.; et al. Production of nuclear transfer-derived swine that express the enhanced green fluorescent protein. Anim. Biotechnol. 2001, 12, 173–181. [Google Scholar] [CrossRef]
- Dai, Y.; Vaught, T.D.; Boone, J.; Chen, S.H.; Phelps, C.J.; Ball, S.; Monahan, J.A.; Jobst, P.M.; McCreath, K.J.; Lamborn, A.E.; et al. Targeted disruption of the alpha1,3-galactosyltransferase gene in cloned pigs. Nat. Biotechnol. 2002, 20, 251–255. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Petersen, B. Basics of genome editing technology and its application in livestock species. Reprod. Domest. Anim. 2017, 52, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Teng, F.; Guo, R.; Li, W.; Zhou, Q. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res. 2014, 24, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, K.M.; Lee, K.; Benne, J.A.; Beaton, B.P.; Spate, L.D.; Murphy, S.L.; Samuel, M.S.; Mao, J.; O’Gorman, C.; Walters, E.M.; et al. Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol. Reprod. 2014, 91, 78. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Rieblinger, B.; Hein, R.; Sfriso, R.; Zuber, J.; Fischer, A.; Klinger, B.; Liang, W.; Flisikowski, K.; Kurome, M.; et al. Viable pigs after simultaneous inactivation of porcine MHC class I and three xenoreactive antigen genes GGTA1, CMAH and B4GALNT2. Xenotransplantation 2019, e12560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, L.; Du, Y.; Xie, F.; Li, L.; Liu, Y.; Liu, C.; Wang, S.; Zhang, S.; Huang, X.; et al. Efficient Generation of Gene-Modified Pigs Harboring Precise Orthologous Human Mutation via CRISPR/Cas9-Induced Homology-Directed Repair in Zygotes. Hum. Mutat. 2016, 37, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Jin, Q.; Ruan, D.; Yang, Y.; Liu, Q.; Wu, H.; Zhou, Z.; Ouyang, Z.; Liu, Z.; Zhao, Y.; et al. Cre-dependent Cas9-expressing pigs enable efficient in vivo genome editing. Genome Res. 2017, 27, 2061–2071. [Google Scholar] [CrossRef]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Schonhuber, N.; Seidler, B.; Schuck, K.; Veltkamp, C.; Schachtler, C.; Zukowska, M.; Eser, S.; Feyerabend, T.B.; Paul, M.C.; Eser, P.; et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med. 2014, 20, 1340–1347. [Google Scholar] [CrossRef]
- Li, S.; Flisikowska, T.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; Saur, D.; Kind, A.; Wolf, E.; Flisikowski, K.; Schnieke, A. Dual fluorescent reporter pig for Cre recombination: Transgene placement at the ROSA26 locus. PLoS ONE 2014, 9, e102455. [Google Scholar] [CrossRef]
- Grossi, A.B.; Hyttel, P.; Jensen, H.E.; Leifsson, P.S. Porcine melanotic cutaneous lesions and lymph nodes: Immunohistochemical differentiation of melanocytes and melanophages. Vet. Pathol. 2015, 52, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Seaton, M.; Hocking, A.; Gibran, N.S. Porcine models of cutaneous wound healing. ILAR J. 2015, 56, 127–138. [Google Scholar] [CrossRef]
- Kragh, P.M.; Nielsen, A.L.; Li, J.; Du, Y.; Lin, L.; Schmidt, M.; Bogh, I.B.; Holm, I.E.; Jakobsen, J.E.; Johansen, M.G.; et al. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res 2009, 18, 545–558. [Google Scholar] [CrossRef]
- Jakobsen, J.E.; Johansen, M.G.; Schmidt, M.; Liu, Y.; Li, R.; Callesen, H.; Melnikova, M.; Habekost, M.; Matrone, C.; Bouter, Y.; et al. Expression of the Alzheimer’s Disease Mutations AbetaPP695sw and PSEN1M146I in Double-Transgenic Gottingen Minipigs. J. Alzheimers Dis. 2016, 53, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.T.; Wang, X.J.; Rohret, J.A.; Struzynski, J.T.; Merricks, E.P.; Bellinger, D.A.; Rohret, F.A.; Nichols, T.C.; Rogers, C.S. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan miniature pigs. PLoS ONE 2014, 9, e93457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Ouyang, H.; Wang, Y.; Pang, D.; Cong, N.X.; Wang, T.; Leng, B.; Li, D.; Li, X.; Wu, R.; et al. Characterization of a hypertriglyceridemic transgenic miniature pig model expressing human apolipoprotein CIII. FEBS J. 2012, 279, 91–99. [Google Scholar] [CrossRef]
- Renner, S.; Fehlings, C.; Herbach, N.; Hofmann, A.; von Waldthausen, D.C.; Kessler, B.; Ulrichs, K.; Chodnevskaja, I.; Moskalenko, V.; Amselgruber, W.; et al. Glucose intolerance and reduced proliferation of pancreatic beta-cells in transgenic pigs with impaired glucose-dependent insulinotropic polypeptide function. Diabetes 2010, 59, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Renner, S.; Braun-Reichhart, C.; Blutke, A.; Herbach, N.; Emrich, D.; Streckel, E.; Wunsch, A.; Kessler, B.; Kurome, M.; Bahr, A.; et al. Permanent neonatal diabetes in INS(C94Y) transgenic pigs. Diabetes 2013, 62, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Klymiuk, N.; Blutke, A.; Graf, A.; Krause, S.; Burkhardt, K.; Wuensch, A.; Krebs, S.; Kessler, B.; Zakhartchenko, V.; Kurome, M.; et al. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum. Mol. Genet. 2013, 22, 4368–4382. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef] [Green Version]
- Schook, L.B.; Collares, T.V.; Darfour-Oduro, K.A.; De, A.K.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K. Unraveling the swine genome: Implications for human health. Annu. Rev. Anim. Biosci. 2015, 3, 219–244. [Google Scholar] [CrossRef] [Green Version]
- Adam, S.J.; Rund, L.A.; Kuzmuk, K.N.; Zachary, J.F.; Schook, L.B.; Counter, C.M. Genetic induction of tumorigenesis in swine. Oncogene 2007, 26, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Saalfrank, A.; Janssen, K.P.; Ravon, M.; Flisikowski, K.; Eser, S.; Steiger, K.; Flisikowska, T.; Muller-Fliedner, P.; Schulze, E.; Bronner, C.; et al. A porcine model of osteosarcoma. Oncogenesis 2016, 5, e210. [Google Scholar] [CrossRef] [PubMed]
- Rubio, R.; García-Castro, J.; Gutiérrez-Aranda, I.; Paramio, J.; Santos, M.; Catalina, P.; Leone, P.E.; Menendez, P.; Rodríguez, R. Deficiency in p53 but not Retinoblastoma Induces the Transformation of Mesenchymal Stem Cells In vitro and Initiates Leiomyosarcoma In vivo. Cancer Res. 2010, 70, 4185–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, H.; Nagai, T.; Harasawa, R.; Yamagami, T.; Takahashi, J.; Ishikawa, K.-I.; Nomura, N.; Nagashima, H. Production of Transgenic Pig Carrying MMTV/v-Ha-ras. J. Reprod. Dev. 1999, 45, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Li, J.; Liu, Y.; Lin, L.; Du, Y.; Li, S.; Yang, H.; Vajta, G.; Callesen, H.; Bolund, L.; et al. High efficiency of BRCA1 knockout using rAAV-mediated gene targeting: Developing a pig model for breast cancer. Transgenic Res. 2011, 20, 975–988. [Google Scholar] [CrossRef]
- Flisikowska, T.; Merkl, C.; Landmann, M.; Eser, S.; Rezaei, N.; Cui, X.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; Wieland, H.; et al. A porcine model of familial adenomatous polyposis. Gastroenterology 2012, 143, 1173–1175. [Google Scholar] [CrossRef]
- Tan, W.; Carlson, D.F.; Lancto, C.A.; Garbe, J.R.; Webster, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc. Natl. Acad. Sci. USA 2013, 110, 16526–16531. [Google Scholar] [CrossRef] [Green Version]
- Callesen, M.M.; Árnadóttir, S.S.; Lyskjaer, I.; Ørntoft, M.W.; Høyer, S.; Dagnaes-Hansen, F.; Liu, Y.; Li, R.; Callesen, H.; Rasmussen, M.H.; et al. A genetically inducible porcine model of intestinal cancer. Mol. Oncol. 2017, 11, 1616–1629. [Google Scholar] [CrossRef] [Green Version]
- Leuchs, S.; Saalfrank, A.; Merkl, C.; Flisikowska, T.; Edlinger, M.; Durkovic, M.; Rezaei, N.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; et al. Inactivation and inducible oncogenic mutation of p53 in gene targeted pigs. PLoS ONE 2012, 7, e43323. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Edlinger, M.; Saalfrank, A.; Flisikowski, K.; Tschukes, A.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; Saur, D.; Kind, A.; et al. Viable pigs with a conditionally-activated oncogenic KRAS mutation. Transgenic Res. 2015, 24, 509–517. [Google Scholar] [CrossRef]
- Schook, L.B.; Collares, T.V.; Hu, W.; Liang, Y.; Rodrigues, F.M.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K.; Singh, K.; Wells, K.D.; et al. A Genetic Porcine Model of Cancer. PLoS ONE 2015, 10, e0128864. [Google Scholar] [CrossRef]
- Principe, D.R.; Overgaard, N.H.; Park, A.J.; Diaz, A.M.; Torres, C.; McKinney, R.; Dorman, M.J.; Castellanos, K.; Schwind, R.; Dawson, D.W.; et al. KRAS(G12D) and TP53(R167H) Cooperate to Induce Pancreatic Ductal Adenocarcinoma in Sus scrofa Pigs. Sci. Rep. 2018, 8, 12548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelsen, M.F.; Callesen, M.M.; Ostergaard, T.S.; Liu, Y.; Li, R.; Callesen, H.; Dagnaes-Hansen, F.; Hamilton-Dutoit, S.; Jakobsen, J.E.; Thomsen, M.K. Pancreas specific expression of oncogenes in a porcine model. Transgenic Res 2017, 26, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Sieren, J.C.; Meyerholz, D.K.; Wang, X.-J.; Davis, B.T.; Newell, J.D., Jr.; Hammond, E.; Rohret, J.A.; Rohret, F.A.; Struzynski, J.T.; Goeken, J.A.; et al. Development and translational imaging of a TP53 porcine tumorigenesis model. J. Clin. Investig. 2014, 124, 4052–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCalla-Martin, A.C.; Chen, X.; Linder, K.E.; Estrada, J.L.; Piedrahita, J.A. Varying phenotypes in swine versus murine transgenic models constitutively expressing the same human Sonic hedgehog transcriptional activator, K5-HGLI2ΔN. Transgenic Res. 2010, 19, 869–887. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.H.; Anders, C.K.; Litton, J.K.; Ruddy, K.J.; Bleyer, A. Breast cancer in adolescents and young adults. Pediatric Blood Cancer 2018, 65, e27397. [Google Scholar] [CrossRef]
- Brenner, D.R.; Brockton, N.T.; Kotsopoulos, J.; Cotterchio, M.; Boucher, B.A.; Courneya, K.S.; Knight, J.A.; Olivotto, I.A.; Quan, M.L.; Friedenreich, C.M. Breast cancer survival among young women: A review of the role of modifiable lifestyle factors. Cancer Causes Control 2016, 27, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast cancer in young women: An overview. Updates Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef]
- De Silva, S.; Tennekoon, K.H.; Karunanayake, E.H. Overview of the genetic basis toward early detection of breast cancer. Breast Cancer 2019, 11, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Gewefel, H.; Salhia, B. Breast cancer in adolescent and young adult women. Clin. Breast Cancer 2014, 14, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [Green Version]
- Donninger, H.; Hobbing, K.; Schmidt, M.L.; Walters, E.; Rund, L.; Schook, L.; Clark, G.J. A porcine model system of BRCA1 driven breast cancer. Front. Genet. 2015, 6, 269. [Google Scholar] [CrossRef] [Green Version]
- COSMIC Database. 2019. Available online: Cancer.sanger.ac.uk (accessed on 17 December 2019).
- Campos, F.G. Colorectal cancer in young adults: A difficult challenge. World J. Gastroenterol. 2017, 23, 5041–5044. [Google Scholar] [CrossRef]
- Siegel, R.L.; Fedewa, S.A.; Anderson, W.F.; Miller, K.D.; Ma, J.; Rosenberg, P.S.; Jemal, A. Colorectal Cancer Incidence Patterns in the United States, 1974–2013. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Connell, L.C.; Mota, J.M.; Braghiroli, M.I.; Hoff, P.M. The Rising Incidence of Younger Patients with Colorectal Cancer: Questions about Screening, Biology, and Treatment. Curr. Treat. Options Oncol. 2017, 18, 23. [Google Scholar] [CrossRef]
- Bailey, C.E.; Hu, C.Y.; You, Y.N.; Bednarski, B.K.; Rodriguez-Bigas, M.A.; Skibber, J.M.; Cantor, S.B.; Chang, G.J. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975–2010. JAMA Surg. 2015, 150, 17–22. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Rowan, A.J.; Lamlum, H.; Ilyas, M.; Wheeler, J.; Straub, J.; Papadopoulou, A.; Bicknell, D.; Bodmer, W.F.; Tomlinson, I.P. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. USA 2000, 97, 3352–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. Wnt Signaling and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, A.H.; Cheng, K.H.; Wong, A.S.; Ng, S.S.; Ma, B.B.; Chan, C.M.; Tsui, N.B.; Chan, L.W.; Yung, B.Y.; Wong, S.C. Current and future molecular diagnostics in colorectal cancer and colorectal adenoma. World J. Gastroenterol. 2014, 20, 3847–3857. [Google Scholar] [CrossRef]
- Mori, Y.; Nagse, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Cheadle, J.P.; Krawczak, M.; Thomas, M.W.; Hodges, A.K.; Al-Tassan, N.; Fleming, N.; Sampson, J.R. Different combinations of biallelic APC mutation confer different growth advantages in colorectal tumours. Cancer Res. 2002, 62, 363–366. [Google Scholar]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Half, E.; Bercovich, D.; Rozen, P. Familial adenomatous polyposis. Orphanet J. Rare Dis. 2009, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Zeineldin, M.; Neufeld, K.L. Understanding phenotypic variation in rodent models with germline Apc mutations. Cancer Res. 2013, 73, 2389–2399. [Google Scholar] [CrossRef] [Green Version]
- Jackstadt, R.; Sansom, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Tetteh, P.W.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; Morsink, F.; Farin, H.; van Es, J.H.; Offerhaus, G.J.; Clevers, H. Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc. Natl. Acad. Sci. USA 2016, 113, 11859–11864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Tamai, Y.; Horiike, S.; Oshima, M.; Taketo, M.M. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Δ716 Cdx2+/− compound mutant mice. Nat. Genet. 2003, 35, 323–330. [Google Scholar] [CrossRef]
- Flisikowska, T.; Stachowiak, M.; Xu, H.; Wagner, A.; Hernandez-Caceres, A.; Wurmser, C.; Perleberg, C.; Pausch, H.; Perkowska, A.; Fischer, K.; et al. Porcine familial adenomatous polyposis model enables systematic analysis of early events in adenoma progression. Sci. Rep. 2017, 7, 6613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.L.; Wang, P.; Lu, M.Z.; Zhang, S.D.; Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019, 17, 4487–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachowiak, M.; Flisikowska, T.; Bauersachs, S.; Perleberg, C.; Pausch, H.; Switonski, M.; Kind, A.; Saur, D.; Schnieke, A.; Flisikowski, K. Altered microRNA profiles during early colon adenoma progression in a porcine model of familial adenomatous polyposis. Oncotarget 2017, 8, 96154–96160. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Rogalla, S.; Flisikowski, K.; Gorpas, D.; Mayer, A.T.; Flisikowska, T.; Mandella, M.J.; Ma, X.; Casey, K.M.; Felt, S.A.; Saur, D.; et al. Biodegradable Fluorescent Nanoparticles for Endoscopic Detection of Colorectal Carcinogenesis. Adv. Funct. Mater. 2019, 1904992. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Quante, A.S.; Ming, C.; Rottmann, M.; Engel, J.; Boeck, S.; Heinemann, V.; Westphalen, C.B.; Strauch, K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016, 5, 2649–2656. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Adsay, N.V.; Albores–Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Klöppel, G.; Longnecker, D.S.; et al. Pancreatic Intraepithelial Neoplasia: A New Nomenclature and Classification System for Pancreatic Duct Lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and repair of the exocrine pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.M.M.; Sancho, R.; Messal, H.A.; Nye, E.; Spencer-Dene, B.; Stone, R.K.; Stamp, G.; Rosewell, I.; Quaglia, A.; Behrens, A. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep. 2017, 21, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, J.; Yokoyama, Y.; Kokuryo, T.; Ebata, T.; Nagino, M. Cells of origin of pancreatic neoplasms. Surg. Today 2018, 48, 9–17. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Yokota, T.; Takano, S.; Yoshitomi, H.; Kagawa, S.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Suzuki, D.; Sakai, N.; Nojima, H.; et al. Successful treatment of a locally advanced unresectable pancreatic cancer patient with interstitial pneumonitis by conversion surgery following gemcitabine plus nab-paclitaxel chemotherapy: A case report. Mol. Clin. Oncol. 2019, 10, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- Pedersen, K.; Bilal, F.; Bernadó Morales, C.; Salcedo, M.T.; Macarulla, T.; Massó-Vallés, D.; Mohan, V.; Vivancos, A.; Carreras, M.J.; Serres, X.; et al. Pancreatic cancer heterogeneity and response to Mek inhibition. Oncogene 2017, 36, 5639–5647. [Google Scholar] [CrossRef] [PubMed]
- Biancur, D.E.; Kimmelman, A.C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F., III; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, N.H.; Principe, D.R.; Schachtschneider, K.M.; Jakobsen, J.T.; Rund, L.A.; Grippo, P.J.; Schook, L.B.; Jungersen, G. Genetically Induced Tumors in the Oncopig Model Invoke an Antitumor Immune Response Dominated by Cytotoxic CD8β+ T Cells and Differentiated γδ T Cells Alongside a Regulatory Response Mediated by FOXP3+ T Cells and Immunoregulatory Molecules. Front. Immunol. 2018, 9, 1301. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Remmers, N.; Cox, J.L.; Grunkemeyer, J.A.; Aravind, S.; Arkfeld, C.K.; Hollingsworth, M.A.; Carlson, M.A. Generation of tumorigenic porcine pancreatic ductal epithelial cells: Toward a large animal model of pancreatic cancer. bioRxiv 2018, 267112. [Google Scholar] [CrossRef] [Green Version]
- Bailey, K.L.; Carlson, M.A. Porcine Models of Pancreatic Cancer. Front. Oncol. 2019, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, K.A.; Barkauskas, D.A.; Krailo, M.D.; Meyers, P.A.; Schwartz, C.L.; Ebb, D.H.; Seibel, N.L.; Grier, H.E.; Gorlick, R.; Marina, N. Outcome for adolescent and young adult patients with osteosarcoma: A report from the Children’s Oncology Group. Cancer 2012, 118, 4597–4605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ognjanovic, S.; Olivier, M.; Bergemann, T.L.; Hainaut, P. Sarcomas in TP53 germline mutation carriers: A review of the IARC TP53 database. Cancer 2012, 118, 1387–1396. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Hansen, M.F.; Koufos, A.; Gallie, B.L.; Phillips, R.A.; Fodstad, O.; Brogger, A.; Gedde-Dahl, T.; Cavenee, W.K. Osteosarcoma and retinoblastoma: A shared chromosomal mechanism revealing recessive predisposition. Proc. Natl. Acad. Sci. USA 1985, 82, 6216–6220. [Google Scholar] [CrossRef] [Green Version]
- Bridge, J.A.; Nelson, M.; McComb, E.; McGuire, M.H.; Rosenthal, H.; Vergara, G.; Maale, G.E.; Spanier, S.; Neff, J.R. Cytogenetic findings in 73 osteosarcoma specimens and a review of the literature. Cancer Genet. Cytogenet. 1997, 95, 74–87. [Google Scholar] [CrossRef]
- Selvarajah, S.; Yoshimoto, M.; Ludkovski, O.; Park, P.C.; Bayani, J.; Thorner, P.; Maire, G.; Squire, J.A.; Zielenska, M. Genomic signatures of chromosomal instability and osteosarcoma progression detected by high resolution array CGH and interphase FISH. Cytogenet. Genome Res. 2008, 122, 5–15. [Google Scholar] [CrossRef]
- Plummer, S.J.; Santibanez-Koref, M.; Kurosaki, T.; Liao, S.; Noble, B.; Fain, P.R.; Anton-Culver, H.; Casey, G. A germline 2.35 kb deletion of p53 genomic DNA creating a specific loss of the oligomerization domain inherited in a Li-Fraumeni syndrome family. Oncogene 1994, 9, 3273–3280. [Google Scholar] [PubMed]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohseny, A.B.; Tieken, C.; van der Velden, P.A.; Szuhai, K.; de Andrea, C.; Hogendoorn, P.C.; Cleton-Jansen, A.M. Small deletions but not methylation underlie CDKN2A/p16 loss of expression in conventional osteosarcoma. Genes Chromosomes Cancer 2010, 49, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef]
- Castillo-Tandazo, W.; Mutsaers, A.J.; Walkley, C.R. Osteosarcoma in the Post Genome Era: Preclinical Models and Approaches to Identify Tractable Therapeutic Targets. Curr. Osteoporos. Rep. 2019, 17, 343–352. [Google Scholar] [CrossRef]
- Walia, M.K.; Castillo-Tandazo, W.; Mutsaers, A.J.; Martin, T.J.; Walkley, C.R. Murine models of osteosarcoma: A piece of the translational puzzle. J. Cell Biochem. 2018, 119, 4241–4250. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Quist, T.; Jin, H.; Zhu, J.F.; Smith-Fry, K.; Capecchi, M.R.; Jones, K.B. The impact of osteoblastic differentiation on osteosarcomagenesis in the mouse. Oncogene 2015, 34, 4278–4284. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, A.J.; Ng, A.J.; Baker, E.K.; Russell, M.R.; Chalk, A.M.; Wall, M.; Liddicoat, B.J.; Ho, P.W.; Slavin, J.L.; Goradia, A.; et al. Modeling distinct osteosarcoma subtypes in vivo using Cre:lox and lineage-restricted transgenic shRNA. Bone 2013, 55, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, M.V.; Ghivizzani, S.C.; Gibbs, C.P. Animal models in osteosarcoma. Front. Oncol. 2014, 4, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Li, X.; Liu, P.; Li, M.; Luo, F. Patient-derived xenograft mouse models: A high fidelity tool for individualized medicine. Oncol. Lett. 2019, 17, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Yada, E.; Wada, S.; Yoshida, S.; Sasada, T. Use of patient-derived xenograft mouse models in cancer research and treatment. Future Sci. OA 2018, 4, Fso271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murayama, T.; Gotoh, N. Patient-Derived Xenograft Models of Breast Cancer and Their Application. Cells 2019, 8, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, I.; Chicote, I.; Tenbaum, S.P.; Arques, O.; Herance, J.R.; Gispert, J.D.; Jimenez, J.; Landolfi, S.; Caci, K.; Allende, H.; et al. A personalized preclinical model to evaluate the metastatic potential of patient-derived colon cancer initiating cells. Clin. Cancer Res. 2013, 19, 6787–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, E.; Hong, S.M.; Yoo, H.J.; Kim, M.B.; Won, J.S.; An, S.; Shim, I.K.; Chang, S.; Hoffman, R.M.; Kim, S.C. Genetic and metabolic comparison of orthotopic and heterotopic patient-derived pancreatic-cancer xenografts to the original patient tumors. Oncotarget 2018, 9, 7867–7881. [Google Scholar] [CrossRef]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Mukae, Y.; Kitsuka, T.; Arai, K.; Nakamura, A.; Uchihashi, K.; Toda, S.; Matsubayashi, K.; Oyama, J.-I.; Node, K.; et al. Development of an immunodeficient pig model allowing long-term accommodation of artificial human vascular tubes. Nat. Commun. 2019, 10, 2244. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Iwamoto, M.; Saito, Y.; Fuchimoto, D.; Sembon, S.; Suzuki, M.; Mikawa, S.; Hashimoto, M.; Aoki, Y.; Najima, Y.; et al. Il2rg Gene-Targeted Severe Combined Immunodeficiency Pigs. Cell Stem Cell 2012, 10, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.T.; Cho, B.; Ryu, J.; Ray, C.; Lee, E.J.; Yun, Y.J.; Ahn, S.; Lee, J.; Ji, D.Y.; Jue, N.; et al. Biallelic modification of IL2RG leads to severe combined immunodeficiency in pigs. Reprod. Biol. Endocrinol. 2016, 14, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Nakano, K.; Matsunari, H.; Matsuda, T.; Maehara, M.; Kanai, T.; Kobayashi, M.; Matsumura, Y.; Sakai, R.; Kuramoto, M.; et al. Generation of interleukin-2 receptor gamma gene knockout pigs from somatic cells genetically modified by zinc finger nuclease-encoding mRNA. PLoS ONE 2013, 8, e76478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Guo, X.; Fan, N.; Song, J.; Zhao, B.; Ouyang, Z.; Liu, Z.; Zhao, Y.; Yan, Q.; Yi, X.; et al. RAG1/2 Knockout Pigs with Severe Combined Immunodeficiency. J. Immunol. 2014, 193, 1496–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Kwon, D.N.; Ezashi, T.; Choi, Y.J.; Park, C.; Ericsson, A.C.; Brown, A.N.; Samuel, M.S.; Park, K.W.; Walters, E.M.; et al. Engraftment of human iPS cells and allogeneic porcine cells into pigs with inactivated RAG2 and accompanying severe combined immunodeficiency. Proc. Natl. Acad. Sci. USA 2014, 111, 7260–7265. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Sendai, Y.; Yamazaki, S.; Seki-Soma, M.; Hirose, K.; Watanabe, M.; Fukawa, K.; Nakauchi, H. Generation of recombination activating gene-1-deficient neonatal piglets: A model of T and B cell deficient severe combined immune deficiency. PLoS ONE 2014, 9, e113833. [Google Scholar] [CrossRef]
- Suzuki, S.; Iwamoto, M.; Hashimoto, M.; Suzuki, M.; Nakai, M.; Fuchimoto, D.; Sembon, S.; Eguchi-Ogawa, T.; Uenishi, H.; Onishi, A. Generation and characterization of RAG2 knockout pigs as animal model for severe combined immunodeficiency. Vet. Immunol. Immunopathol. 2016, 178, 37–49. [Google Scholar] [CrossRef]
- Fisher, J.E.; Lillegard, J.B.; McKenzie, T.J.; Rodysill, B.R.; Wettstein, P.J.; Nyberg, S.L. In utero transplanted human hepatocytes allow postnatal engraftment of human hepatocytes in pigs. Liver Transplant. 2013, 19, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Sinkora, M.; Sinkora, J.; Rehakova, Z.; Butler, J.E. Early ontogeny of thymocytes in pigs: Sequential colonization of the thymus by T cell progenitors. J. Immunol. 2000, 165, 1832–1839. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Cancer | Genetic Modification | Generated By | Comments | Reference |
|---|---|---|---|---|
| Breast Cancer | MMTV/v-Ha-ras (transgene) | Microinjection | No phenotype | [73] |
| Heterozygous BRCA1 knockout (endogenous gene) | Gene targeting via AAV + SCNT | No survival of born piglets | [74] | |
| Colorectal Cancer | Heterozygous APC1311 truncation mutation (endogenous gene) | Gene targeting + SCNT | Colonic polyposis | [75] |
| Heterozygous APC902 truncation mutation (endogenous gene) | TALENs + Chromatin transfer | No phenotype | [76] | |
| Flp-inducible KRASG12D + cMYC+SV40LT (transgenes) | Random integration + SCNT | Villin-driven; Duodenal carcinoma | [77] | |
| Pancreatic Cancer | Cre-inducible TP53R167H + KRASG12D mutation (endogenous genes) | Gene targeting + SCNT | Pancreas-specific activation intended | [78,79] |
| Cre-inducible TP53R167H + KRASG12D mutation (transgenes) | Random integration + SCNT | AdCre delivery into duct led to tumor formation | [80,81] | |
| Flp-inducible KRASG12D + cMYC+SV40LT (transgenes) | Random integration + SCNT | Pdx1-driven; Hyperplastic foci of acinar cells | [82] | |
| Osteosarcoma | Hetero- and homozygous knockout of TP53 major transcript (endogenous gene) | Gene targeting + SCNT | OS primarily affecting long bones | [71] |
| Homozygous TP53R167H mutation (endogenous gene) | Gene targeting via AAV + SCNT | Various lesions, e.g., osteogenic tumors, lymphomas and renal tumors | [83] | |
| Other Cancers | Human Gli2 transcriptional activator K5-hGli2ΔN (transgene) | Random integration + SCNT | Basal cell carcinoma-like lesions; infection | [84] |
| Cre-inducible TP53R167H + KRASG12D mutation (endogenous genes) | Gene targeting + SCNT | Suitable for diverse cancers, e.g., lung cancer | [78,79] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalla, D.; Kind, A.; Schnieke, A. Genetically Engineered Pigs to Study Cancer. Int. J. Mol. Sci. 2020, 21, 488. https://doi.org/10.3390/ijms21020488
Kalla D, Kind A, Schnieke A. Genetically Engineered Pigs to Study Cancer. International Journal of Molecular Sciences. 2020; 21(2):488. https://doi.org/10.3390/ijms21020488
Chicago/Turabian StyleKalla, Daniela, Alexander Kind, and Angelika Schnieke. 2020. "Genetically Engineered Pigs to Study Cancer" International Journal of Molecular Sciences 21, no. 2: 488. https://doi.org/10.3390/ijms21020488
APA StyleKalla, D., Kind, A., & Schnieke, A. (2020). Genetically Engineered Pigs to Study Cancer. International Journal of Molecular Sciences, 21(2), 488. https://doi.org/10.3390/ijms21020488